Inflammation and Oxidative-Stress Pathways Are Associated with Idiopathic Sudden Hearing Loss: A Genome-Wide Association Study in 15,494 Japanese Individuals
Ryosuke Kitoh, Shin-Ya Nishio, Yutaka Takumi, Shin-ichi Usami

TL;DR
This study identifies genetic loci linked to sudden hearing loss in Japanese individuals, suggesting inflammation and oxidative stress play a role.
Contribution
The study provides the first genome-wide association evidence for immune-inflammatory and stress-related pathways in idiopathic sudden hearing loss.
Findings
Eight loci reached genome-wide significance, including FHIT and others near LHX2 and TRMT1L.
MAGMA identified eight genes associated with iSSNHL at FDR < 0.05.
Findings suggest immune-inflammatory and cellular stress mechanisms contribute to iSSNHL susceptibility.
Abstract
The etiology of idiopathic sudden sensorineural hearing loss (iSSNHL) remains unclear, and genome-wide genetic evidence is limited. We conducted a multicenter Japanese case–control genome-wide association study including 192 clinically defined iSSNHL cases and 15,302 controls aged ≥80 years without a history of hearing loss. After cross-platform SNP harmonization and imputation (Eagle/Minimac4), association testing was performed using dosage-based logistic regression in PLINK 2.0, adjusting for sex and principal components (PC1–PC10). Gene- and pathway-level analyses were conducted using MAGMA and the PANTHER overrepresentation test. Genomic inflation was modest (λ_GC = 1.04). Eight loci reached genome-wide significance (p < 5 × 10−8), led by FHIT, with additional loci near LHX2, TRMT1L, MEGF10, SPATS1, SAMD5, MYT1L, and ID4; 21 loci met the suggestive threshold (p < 1 × 10−6). MAGMA…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
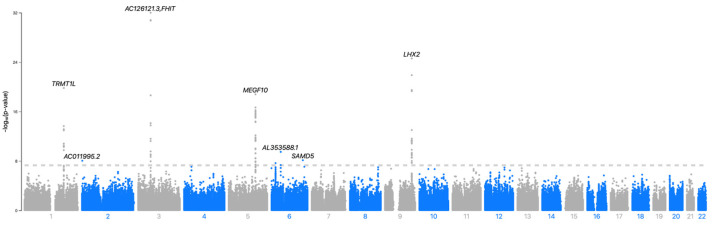 Figure 1
Figure 1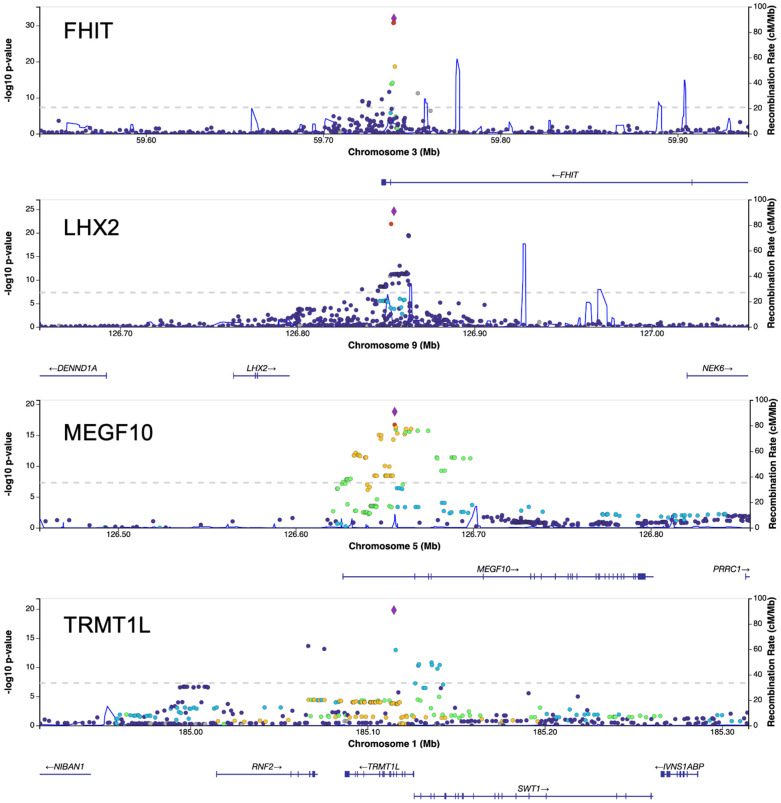 Figure 2
Figure 2- —Health and Labour Sciences Research Grants for Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labour and Welfare, Japan
- —JSPS KAKENHI
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVestibular and auditory disorders · Hearing, Cochlea, Tinnitus, Genetics · Neuroinflammation and Neurodegeneration Mechanisms
1. Introduction
Idiopathic sudden sensorineural hearing loss (iSSNHL) is a rapidly developing sensorineural hearing loss that occurs within 72 h. International epidemiological studies estimate an annual incidence of 5–27 per 100,000 people [1], whereas a nationwide Japanese epidemiological survey reported an incidence of 60.9 (95% CI 57.6–64.2) per 100,000 people [2]. Although local circulatory disturbance, viral infection, autoimmunity, and oxidative stress have been proposed as etiologies, the definitive mechanism remains unclear [3,4,5].
With respect to genetic contributions, previous studies have mainly employed candidate-gene association approaches. A recent systematic review summarizing 47 studies (5230 patients) and 68 genes reported associations with single nucleotide polymorphisms (SNPs) in several pathways related to coagulation/thrombosis (e.g., F2, F5, MTHFR, ITGA2/ITGB3), inflammation and immunity (e.g., IL-1, IL-6, TNF, ICAM1), oxidative stress and free-radical processing (e.g., SOD1, GSTP1, GPX, GSR, NOS3), as well as genes located in or near the HLA region [6].
Consistent with these observations, we previously conducted a two-stage candidate-gene association study using the Intractable Inner Ear Disease Gene Bank and found that SOD1 rs4998557, together with GSTP1 and PRKCH SNPs, may contribute to susceptibility to SSNHL, particularly among patients with hearing loss ≥60 dB and tinnitus [7]. Using the same cohort, we further demonstrated that SNPs in oxidative-stress and vascular genes (GSR rs2251780/rs3779647 and NOS3 rs1799983) and in the glucocorticoid receptor gene NR3C1 rs4912910 were associated with differences in hearing recovery after standardized corticosteroid therapy, suggesting that germline variations may influence not only disease risk but also prognosis and treatment response [8]. Nevertheless, most candidate-gene association studies, including our own, have been based on relatively small case–control series and have evaluated a limited number of loci, and the overall reproducibility of individual associations has been modest because of insufficient statistical power, multiple testing, population differences, and patient-recruitment bias [6].
As iSSNHL is likely a polygenic disorder to which many small-effect SNPs contribute, a hypothesis-free genome-wide association study (GWAS) followed by gene-level analyses (e.g., MAGMA) and pathway-level interpretation (pathway enrichment) is a more suitable approach than candidate-gene-based analysis. Indeed, while large-scale GWASs have been performed for age-related hearing loss and self-reported “hearing difficulty”, rigorously defined iSSNHL has rarely been subjected to a large GWAS because of its lower incidence [6].
In this study, we conducted a GWAS of iSSNHL using a multicenter Japanese cohort. By connecting single-nucleotide polymorphism (SNP)-level results with gene- and biological pathway-level analyses, we aimed to provide genome-wide evidence and generate testable hypotheses regarding the molecular mechanisms underlying disease susceptibility.
2. Results
2.1. Participant Characteristics
This study analyzed an iSSNHL patient cohort comprising 192 samples from 11 facilities. Age at onset was available for 181 cases and unavailable for 11 cases: the mean age at onset for the 181 cases was 56.1 ± 15.4 years (range, 9–86 years; median, 59 years (interquartile range [IQR] 46–67)). The cohort included 109 males and 83 females. The mean hearing level (average of 250, 500, 1000, 2000, and 4000 Hz) in the affected ear before treatment was as follows: <40 dB in 19 cases, ≥40 dB and <60 dB in 41 cases, ≥60 dB and <90 dB in 78 cases, and ≥90 dB in 54 cases.
The final dataset used for association testing comprised 15,494 individuals (192 cases and 15,302 controls), selected under the criteria described in the Methods.
2.2. GWAS Results (SNP-Level Analysis)
Genomic inflation was modest (λ_GC = 1.04), indicating no major systematic bias. We identified eight loci that reached genome-wide significance (p < 5 × 10^−8^) (see Table 1). The rsIDs in Table 1 were unified according to dbSNP (version 155; GRCh37). Only variants without an assigned rsID are shown as “chr:pos (GRCh37)”. Figure 1 shows the genome-wide Manhattan plot, and Figure 2 shows LocusZoom regional association plots for the four top loci: rs6803403 (FHIT, chromosome 3), rs72759216 (LHX2, chromosome 9), rs246887 (MEGF10, chromosome 5), and rs6684586 (TRMT1L, chromosome 1). In addition to the eight genome-wide significant loci, we identified 21 loci that fulfilled the suggestive threshold (5 × 10^−8^ ≤ p < 1 × 10^−6^), as summarized in Table 2.
In each region, the most significant variant (purple diamond) and a cluster of additional variants with high LD also showed strong association (dashed line at p = 5 × 10^−8^), providing the input for the subsequent COJO analyses. The genomic context in the plots is consistent with the annotation in Table 1 (FHIT/TRMT1L/MEGF10: overlap; LHX2: intergenic). For each lead variant, the complete list of genes within ±250 kb together with the overlap/distance labels defined in Methods is provided in Supplementary Table S1. To facilitate interpretation of the effect sizes, dosage-based allele frequencies and dosage-derived minor allele counts for each lead SNP are provided in Supplementary Table S2.
2.3. Conditional and Joint Analysis Results
Using GCTA-COJO (stepwise selection; COJO-SLCT), we identified multiple independent signals at FHIT (three signals), TRMT1L (three signals), and LHX2 (two signals) (Table 3). These remained clearly detectable in excl models; i.e., when conditioning was performed on the other COJO-selected leads while excluding the target lead, supporting their independence. In contrast, MEGF10, ID4, and SPATS1 each harbored a single lead, and the post-all minimum pC did not reach genome-wide significance at these loci (Table 3). SAMD5/MYT1L did not yield independent signals in the selection step (No. selected leads = 0). For TRMT1L, the post-all minimum pC was 5.13 × 10^−8^, close to the genome-wide significance threshold. Given the approximate nature of external LD references, we refrained from over-interpretation.
2.4. Gene-Level Results (MAGMA)
Gene-based testing with MAGMA (16,025 genes) identified eight genes that reached statistical significance after BH–FDR correction (FDR < 0.05; Table 4). FHIT showed the strongest association (p = 2.3 × 10^−30^, FDR = 3.6 × 10^−26^), followed by TRMT1L and MEGF10 (p ≈ 7.2–7.7 × 10^−16^). RNF2, SWT1, VAMP1, TAPBPL and C9orf3 also remained significant after correction. Notably, FHIT, TRMT1L and MEGF10 overlapped the top SNP-level loci, whereas RNF2, SWT1, VAMP1, TAPBPL and C9orf3 emerged only at the gene level.
2.5. Pathway Enrichment Results (PANTHER)
Based on the GWAS results, we constructed a GWAS-derived gene set for pathway analysis. For loci that reached genome-wide significance, genes located within a ±250-kb window around each lead SNP were taken from the positional gene lists in Supplementary Table S1 and cleaned according to the procedure in Methods (Section 4.9), leaving 28 genes. For loci that met the suggestive threshold in Table 2, we selected the nearest gene(s) at each independent locus and, after the same cleaning, obtained 16 genes summarized in Supplementary Table S3. Together, these 28 and 16 genes formed a 44-gene GWAS-derived “clean set,” which we analyzed with the PANTHER Overrepresentation Test (PANTHER v19.0) using the Homo sapiens genome as the reference list and Fisher’s exact test with Benjamini–Hochberg FDR correction, with UNCLASSIFIED categories excluded.
No pathway reached the predefined significance threshold of FDR < 0.05. Nevertheless, several pathways showed nominal enrichment when ranked by fold enrichment and raw p value (Table 5). Among these, the gonadotropin-releasing hormone receptor pathway and the “inflammation mediated by chemokine and cytokine signaling” pathway were notable. The latter pathway included IFNG, NFKBIE, and ITPR2, which is consistent with a potential contribution of immune-inflammatory mechanisms to iSSNHL susceptibility. As no pathway passed FDR < 0.05, these pathway-level observations should be interpreted as exploratory (hypothesis-generating).
3. Discussion
3.1. Comparison with Previous Studies and the Originality of This Work
Early genetic studies of iSSNHL employed candidate-gene association study-based approaches, focusing on specific genes such as HLA types, inflammatory cytokines (e.g., IL6 and TNF), coagulation-related genes (F2, F5, and MTHFR), and oxidative stress-related genes (e.g., SOD1, GSTP1, GPX1/3, GSR, and PON1), as summarized in a recent systematic review [6]. The reproducibility of these findings has been hindered by a combination of small sample sizes, the burden of multiple comparisons, and population heterogeneity [6]. In contrast, GWASs of hearing-related traits that focused on age-related hearing loss (ARHL), self-reported “hearing difficulty,” tinnitus, and vertigo have been performed previously [9,10,11,12]. More recently, a Mendelian randomization analysis using FinnGen iSSNHL cases explored the causal relationship with inflammation-related SNPs [13]. Nevertheless, case–control GWASs of clinically well-phenotyped iSSNHL, diagnosed under unified criteria and accompanied by detailed clinical information, remain scarce.
The originality of the present study can be summarized as follows:
Definition of phenotype and multi-institutional accrual: iSSNHL samples were collected through the Intractable Inner Ear Diseases Gene Bank Japan, which involves 23 collaborating institutions across Japan under uniform diagnostic criteria; the present GWAS analyzed 192 cases accrued from 11 institutions (including Shinshu University).
Control design: We adopted a “resilient” control phenotype, which was defined as individuals aged ≥80 years at sampling without a history of hearing loss from BBJ, to minimize the risk of future incident iSSNHL cases.
Multilayer interpretation: SNP-level GWAS identified eight genome-wide significant loci (FHIT, LHX2, TRMT1L, MEGF10, SPATS1, SAMD5, MYT1L, and ID4), and gene-based analysis (MAGMA) identified eight genes at FDR < 0.05 (FHIT, TRMT1L, MEGF10, RNF2, SWT1, VAMP1, TAPBPL, and C9orf3). The PANTHER pathway overrepresentation analysis did not yield any pathway passing FDR < 0.05; therefore, pathway-level observations based on nominal p values should be considered exploratory and require replication.
Taken together, this work represents an early data-driven GWAS of clinically well-phenotyped iSSNHL in a Japanese cohort and provides a framework for future replication and functional follow-up. In a cohort of 192 cases and 15,302 controls, we observed SNP-level and gene-level signals consistent with immune-inflammatory and cellular stress–homeostasis mechanisms; however, pathway-level signals from PANTHER were not FDR-significant and are therefore hypothesis-generating. In the SNP-level analysis, rs72759216 near the LHX2 locus was among the strongest associations in our GWAS, even though LHX2 itself did not reach gene-based significance in MAGMA and did not anchor an FDR-significant pathway. LHX2 encodes a LIM-homeodomain transcription factor essential for forebrain development and thalamocortical axonal guidance, including pathways from the auditory thalamus [14]. Its implication in an adult-onset disorder such as iSSNHL is therefore intriguing but should be interpreted cautiously; we consider this a developmental susceptibility hypothesis that will require replication and targeted functional studies to clarify the causal gene and mechanism.
3.2. Two Pathways of the Pathophysiological Model
3.2.1. The Inflammation/Immunity Pathway: Integration of IFNG and the NF-κB Pathway
Integrating the SNP-level GWAS results with MAGMA and PANTHER suggests an inflammation/immunity axis centered on the IFNG, ITPR2, and NFKBIE loci. In our data, lead variants near IFNG and ITPR2 reached the suggestive threshold (p ≤ 1 × 10^−6^), whereas NFKBIE lay within the ±250 kb window of one of the genome-wide significant loci; these genes were included in the GWAS-derived gene set and mapped to the “inflammation mediated by chemokine and cytokine signaling” pathway in PANTHER. However, because no pathway passed FDR < 0.05, this pathway-level interpretation should be considered hypothesis-generating and requires replication.
(1)IFN-γ/Th1 pathway.
In autoimmune sensorineural hearing loss (ASNHL), T cells that react to inner ear antigens produce IFN-γ. This suggests that an acute/subacute Th1-skewed environment can develop in sensorineural hearing loss [15]. In inner ear models, IFN-γ pretreatment activates JAK1/2–STAT1 and increases susceptibility to TNFα toxicity via TNFR1-dependent caspase1. Moreover, IFN-γ and TNFα act synergistically to aggravate cisplatin-induced hair cell injury [16]. Spiral ligament fibrocytes can produce TNFα in response to inflammatory stimuli, suggesting that local cellular constituents may initiate inflammation. The genetic observations near IFNG in our GWAS are compatible with such upstream sensitization mechanisms.
(2)Ca^2+^–NF-κB response.
ITPR2 mediates intracellular Ca^2+^ release and is involved in signal amplification under inflammatory stimulation [17]. NFKBIE restrains NF-κB activation and variations in this brake may shift stress response thresholds. Temporal bone pathology suggests that classical vascular occlusion or membrane rupture alone cannot explain all iSSNHL cases, raising the possibility that pathological activation of cellular stress pathways (including NF-κB) contributes to disease [3]. In mice, NF-κB deficiency is associated with auditory nerve degeneration and exacerbated noise-induced hearing loss [18], and reports variably position NF-κB as both protective and detrimental within cochlear oxidative-stress responses.
(3)Integration and therapeutic implications.
Dexamethasone-mediated oto-protection has been linked to PI3K/Akt/NF-κB signaling [19]; thus, genetic heterogeneity along the NF-κB pathway could underlie differential steroid responsiveness in iSSNHL. In summary, our findings related to the inflammatory pathway suggest the existence of genetically determined thresholds within a cascade comprising IFN-γ-driven sensitization, Ca^2+^ dynamics via ITPR2-mediated amplification, and NF-κB responses modulated by NFKBIE. Prospective stratification combining IFN-γ/TNFα profiles and genotype may help to identify individual inflammatory drivers and response pathways.
3.2.2. The Non-Inflammatory Pathway: Oxidative Stress and Cellular Homeostasis (FHIT/TRMT1L/MEGF10)
Although our focus is on inflammation and immunity, the top signals displaying concordance between GWAS and MAGMA (FHIT, TRMT1L, and MEGF10) support a complementary pathway of cell-intrinsic stress responses and homeostatic maintenance that may operate upstream or downstream of inflammation.
FHIT spans the common fragile site FRA3B and contributes to genome stability by coping with replicative stress and the DNA damage checkpoint/repair process [20]. Loss of FHIT increases replication stress and double-strand breaks, thereby promoting genome instability via dysregulated stress responses [21]. FHIT also cooperates with FDXR (ferredoxin reductase) in mitochondria to regulate apoptosis thresholds under severe oxidative stress [22]. In the inner ear, ischemia/reperfusion and noise exposure elevate lipid peroxidation and increase 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxo-dG), among other markers; thus, the known roles of FHIT in genome stability and modulation of oxidative stress may contribute to interindividual susceptibility to acute threshold shifts [23,24].
TRMT1L participates in tRNA methylation and post-transcriptional modification, and is thereby linked to protein quality control and stress granule dynamics under oxidative stress [25,26]. Similar to FHIT, SNPs in TRMT1L could influence the handling of oxidative stress relevant to iSSNHL onset.
MEGF10 is a receptor that recognizes and engulfs apoptotic cells and cell debris (efferocytosis), acting in concert with opsonins such as C1q and phagocytic “eat me” signals [27]. In the central nervous system, MEGF10 binds C1q to mediate clearance of apoptotic cells by astrocytes [28]. In the mouse cochlea (from P8 to P20), MEGF10 shows relatively higher expression in supporting cells in sc/snRNA-seq datasets (visualized in gEAR) [29]. During hair cell injury in the inner ear, the clearance of dead hair cells/fragments by supporting cells is thought to curb secondary inflammation and help maintain ionic homeostasis [30]. While indirect, these mechanisms suggest that differences in MEGF10 function could modulate iSSNHL susceptibility.
3.3. Clinical Implications
Our GWAS indicates genetic signals spanning inflammation and oxidative-stress responses, which is consistent with the conventional theory that the disease is multifactorial rather than being caused by a single mechanism. Clinically, stratification and treatment choices should consider two pathways: (a) inflammation, and (b) oxidative stress/cellular homeostasis. Recent reviews and clinical studies support the roles of inflammation, metabolic dysregulation, and oxidative stress, and suggest the utility of blood-based markers for prognosis [31,32].
Potential applications include: Stratification by “visualizing” the inflammation pathway in combination with genotype. At initial presentation, low-cost NLR/PLR indices derived from routine blood counts are accumulating evidence of an association with onset and prognosis [33]. IFN-γ and TNFα levels can rise acutely and decline with recovery [34]. Combining such acute phase biomarkers with genotypes (e.g., IFNG/NFKBIE) may enable future risk stratification and therapy selection.
NF-κB functional readouts and oxidative-stress markers. NF-κB is a hub of cochlear inflammatory responses and is activated by noise and other stressors [34,35]. In research settings, peripheral blood mononuclear cell (PBMC) assays involving short-term stimulation followed by p65 (RelA) phosphorylation and NF-κB target gene expression can serve as functional readouts [34]. As steroids classically suppress NF-κB activity [35], pairing NF-κB-related genotypes with PBMC readouts to examine differences in steroid responsiveness is feasible and merits evaluation in prospective studies. Oxidative-stress markers such as 8-OhdG and lipid peroxidation (MDA/TBARS) change in SSNHL and are emphasized in pathophysiological reviews [5,32]. Exploratory use of these markers alongside genotypes (e.g., FHIT/TRMT1L) could support stratification by stress tolerance.
3.4. Strengths and Limitations
Although the case series is moderate in size for GWAS, we leveraged a large control set, achieving a favorable λ_GC = 1.04. However, in a sensitivity analysis requiring Rsq > 0.8, genome-wide significance persisted for FHIT, TRMT1L, MEGF10, and LHX2, while other variants were not significant, and replication is therefore necessary for these. Furthermore, the genes (variants) reaching significance here require functional validation (expression, eQTL, cellular/animal models) to confirm causality. Several lead variants displayed very large ORs with wide confidence intervals, consistent with low allele counts for low-frequency variants (Supplementary Table S2); these estimates should be interpreted cautiously and require replication.
An additional limitation is the extreme age imbalance between cases (predominantly midlife onset) and controls (≥80 years), which is inherent to our “resilient control” design. Although germline genotypes are fixed and do not change with age, using very old controls may introduce residual confounding via age-related survival or birth-cohort effects that can influence allele frequencies. We did not perform age-matched subsampling or include age as a covariate, as age was an explicit component of the control definition; replication in age-matched cohorts and alternative designs will be important.
In addition, our analyses were limited to germline genetic variation and did not assess epigenetic regulation (e.g., DNA methylation and chromatin-level regulatory mechanisms) or gene-environment interactions that may influence iSSNHL susceptibility. Future studies integrating epigenomic and transcriptomic profiling with GWAS will be important to clarify the regulatory mechanisms underlying the implicated inflammatory and oxidative-stress pathways.
4. Materials and Methods
4.1. Study Design and Ethics
This study was a multicenter case–control genome-wide association study. The protocol was approved by the Institutional Review Board of Shinshu University School of Medicine (Approval No. 5528; date of approval: 31 May 2022), and written informed consent was obtained from all participants.
4.2. Cases
Genomic DNA was obtained from peripheral blood samples of patients with iSSNHL who were registered in the Intractable Inner Ear Diseases Gene Bank Japan. This biobank includes samples collected at 23 collaborating research facilities across Japan, as reported previously [7], and contains detailed clinical information, including sex and age at onset, hearing-loss severity, accompanying symptoms (e.g., tinnitus and vertigo), treatment, and outcomes. Of these, 11 facilities (including Shinshu University) contributed the 192 case samples analyzed in the present GWAS.
The diagnostic criteria for SSNHL unified the domestic standards, based on the 2018 edition of the Clinical Practice Guidelines for the Diagnosis and Management of Acute Sensorineural Hearing Loss (Japan Audiological Society; Kanehara & Co., Tokyo, Japan; in Japanese), as well as the revised criteria proposed by the Research Group on Acute Severe Sensorineural Hearing Loss in 2012 [36]. The summary of the specific diagnostic criteria is as follows:
Main symptoms: Sudden onset of sensorineural hearing loss, which is usually severe and of unknown etiology.
For reference: Hearing loss of ≥30 dB over three consecutive frequencies, with the possibility of progressive deterioration within 72 h, is also specified in the guidelines.
4.3. Controls
In case–control studies of iSSNHL, individuals without a prior diagnosis at the time of DNA sampling may still develop iSSNHL later in life. To reduce this potential misclassification, we defined controls as individuals aged ≥80 years at DNA sampling with no history of hearing loss. This design was supported by our nationwide epidemiological survey of >3400 individuals with iSSNHL, in which most cases occurred before age 80 and only 3.1% had onset at age ≥80 years [36]. Accordingly, age was treated as part of the control phenotype definition rather than a covariate in the primary association model.
To obtain a sufficiently large control set meeting these criteria, we used the Biobank Cross Search system to identify eligible samples across Japanese biobanks. BioBank Japan (BBJ) provided the majority of eligible samples. Genotype data for 15,494 BBJ participants who met the above criteria and were enrolled in 47 common disease projects [37] were obtained from the National Bioscience Database Center (NBDC), Japan Science and Technology Agency, via the Japanese Genotype-phenotype Archive (JGA; dataset ID: JGAD000123) after ethical approval and under Data Access Committee-approved data-use agreements (Application ID: JDU000716002) [38]. Following the sample-level QC procedures described below (including missingness filtering, relatedness filtering, and population-structure assessment), 15,302 participants remained and were used as controls in the final GWAS.
4.4. Genotyping, Data Harmonization, Imputation, and Quality Control
4.4.1. Genotyping Arrays
Cases were genotyped using Affymetrix Axiom Japonica Array^®^ v2 (Thermo Fisher Scientific, Waltham, MA, USA) at Toshiba, Co., Ltd. (Tokyo, Japan) according to the manufacturer’s protocol/instructions. Data processing followed StaGen’s (StaGen, Tokyo, Japan) in-house standard operating scripts, in the order below.
4.4.2. Format Conversion and Initial Alignment
The BBJ final reports were converted to PLINK format (final report → tped/tfam → bed/bim/fam). Based on BLAST (2.17.0)-derived coordinates and allele information, all genotypes were unified to the forward strand.
4.4.3. Within-Group QC
SNP-level filters were applied stepwise to cases and controls separately: missingness (option --geno 0.01), minor allele frequency (MAF) ≥ 0.01, and Hardy–Weinberg equilibrium (HWE) p ≥ 1 × 10^−4^ in controls. At the sample level, individuals with missing call rates > 2% were excluded, and outliers in heterozygosity (median ± 4 SD) were removed.
4.4.4. Harmonization (Common SNP Definition)
To avoid inconsistencies between different arrays, a unified SNP identifier was generated: CHR: POS: Minor: Major. Only SNPs shared between cases and controls under this common definition were extracted and merged.
4.4.5. VCF Conversion and Imputation
VCFs for each chromosome from the common SNP set were submitted to the local Michigan Imputation Server (https://imputationserver.sph.umich.edu/ (accessed on 3 January 2026)) for pre-phasing using Eagle v2.4 (https://data.broadinstitute.org/alkesgroup/Eagle/ (accessed on 3 January 2026)), followed by imputation using Minimac4 v4.1.6 (https://genome.sph.umich.edu/wiki/Minimac4 (accessed on 3 January 2026)) with the 1000 Genomes Project Phase 3 East Asian (EAS) reference and GRCh37/hg19 [39].
4.4.6. Post-Imputation QC
From the imputed VCFs, variants with Rsq ≥ 0.3 and MAF > 0.001 were retained for dosage-based analyses, generating the final GWAS input. As the imputation outputs lacked rsIDs, we assigned these from the latest dbSNP chromosome-wise VCF for GRCh37/hg19 (dbSNP version 155 (GRCh37), Release 8 April 2021), as detailed below. Finally, the total number of variants entered into the GWAS (autosomes chr1–22) was 5,187,415.
4.4.7. rsID Assignment
Variants were matched by CHR: POS: REF: ALT in order to assign current, valid dbSNP rsIDs. Ambiguous A/T and C/G alleles were checked to ensure there was no strand discordance with the GRCh37 reference strand. rsIDs were only assigned for multiallelic sites when the REF and ALT allele pair matched exactly. Variants that were not registered in dbSNP or could not be uniquely resolved were not assigned an rsID and were displayed as “chr:pos” (GRCh37).
4.4.8. Display Rules
In the main text and tables, rsIDs are displayed preferentially (e.g., rs123456). Only variants without an assigned rsID are shown as “chr:pos”. For internal procedures such as COJO, the position ID (posID = “CHR:POS”) was used consistently; reader-facing materials include rsIDs in parallel (see Section 4.7 for COJO details).
4.5. Population Structure and Sample QC
From the set of common SNPs, individuals with missing call rates > 2% were excluded, as were heterozygosity outliers (median ± 4 SD). Cryptic relatedness was evaluated using identity-by-descent (IBD) estimates in PLINK, and related individuals were removed using a threshold of PI_HAT > 0.1875. After sample-level QC and population-structure assessment, the final analytic dataset comprised 192 cases and 15,302 controls (N = 15,494). After linkage disequilibrium (LD) pruning (MAF ≥ 0.05, HWE p ≥ 1 × 10^−4^, r^2^ < 0.1), principal component analysis (PCA) was performed using flashPCA v2.1 (https://github.com/gabraham/flashpca (accessed on 2 January 2026)) with outliers (mean ± 6 SD on each PC) excluded [40]. The final regression model included sex and PC1–PC10.
4.6. GWAS (SNP-Level Analysis)
Logistic regression analysis was performed using dosage (DS) from imputed VCFs (Rsq ≥ 0.3, MAF > 0.001) using PLINK v2.0 (https://www.cog-genomics.org/plink/2.0/ (accessed on 2 January 2026)) (generalized linear model module), with case–control status as the outcome, DS as the predictor, and sex plus PC1–PC10 as covariates [41,42]. Genome-wide significance was defined as p = 5 × 10^−8^, and suggestive significance as p = 1 × 10^−6^. The genomic inflation factor λ_GC was computed using autosomal SNPs with Rsq ≥ 0.8 and MAF > 0.01. If a lead SNP overlapped the gene coding sequence, the corresponding gene was annotated as an overlap; otherwise, the nearest gene was assigned based on the shortest distance to a gene-body boundary. Gene boundaries were taken from NCBI37.3.gene.loc (GRCh37/hg19). For positional context, we defined “nearby genes” as those whose gene bodies fall within ±250 kb of each lead variant. Regional association plots were generated with the LocusZoom web portal (built on LocusZoom.js) [43], with 1000 Genomes Phase 3 EAS LD and GRCh37/hg19 coordinates. Each panel displays −log_10_P against the genomic position within a ±200 kb window centered on the lead variant (purple diamond). Dots are color-coded by LD r^2^ with the lead, while gray indicates variants for which no LD information is available. The recombination rate (cM/Mb) is shown in blue. The dashed line denotes p = 5 × 10^−8^ (−log_10_P = 7.30). Age was not included as a covariate in the primary model because controls were deliberately defined as individuals aged ≥80 years (“resilient controls”) to minimize future case misclassification; potential limitations of this design are discussed in the Discussion (Section 3.4).
4.7. Conditional and Joint Analysis (COJO)
For genome-wide significant loci, we evaluated the presence of multiple independent signals using GCTA-COJO v1.94.1 (https://yanglab.westlake.edu.cn/software/gcta/ (accessed on 2 January 2026)). For each region (± 2 Mb around the index SNP), we performed conditional analyses using 1000 Genomes Phase 3 EAS as the linkage disequilibrium (LD) reference (n = 504; PLINK format). SNP IDs were unified as posID = “CHR:POS”, and only variants common to the jma file (summary statistics) and the LD reference were used.
First, stepwise selection of independent lead SNPs was conducted with GCTA-COJO (step-wise selection; COJO-SLCT) using the threshold p < 5 × 10^−8^, collinearity cutoff of 0.9, and a ± 2 Mb window. Next, each selected lead SNP was re-estimated conditioning on the other leads, and regional plots were generated for pre- (unadjusted) and post-excl statuses. Finally, post-all estimates were obtained with GCTA-COJO (conditional model including all selected leads), and the minimum pC within each region was summarized in a table. Visualization was performed using a minimal script to highlight the independent leads with large diamonds and only label the focal lead.
4.8. Gene-Level Analysis (MAGMA)
Gene-level associations were assessed using MAGMA v1.10 (https://ctg.cncr.nl/software/magma (accessed on 2 January 2026)) [44] in the summary-statistics mode. SNPs were mapped to genes using the NCBI37.3.gene.loc definitions (GRCh37/hg19) with a ±0 kb window around the gene body. The SNP-wise multi (“multi”, P_MULTI) gene test was applied, using the 1000 Genomes Project Phase 3 East Asian (EAS) panel as the LD reference. We set the sample size modifier to N = 15,494 (cases + controls). Statistical significance after multiple testing was defined using a Benjamini–Hochberg false discovery rate (BH-FDR) threshold of <0.05 across all autosomal genes tested (n = 16,025).
4.9. Pathway Enrichment Analysis (PANTHER)
We performed a pathway overrepresentation analysis of GWAS-derived loci using PANTHER (http://www.pantherdb.org/) [45]. First, we identified lead SNPs that met either genome-wide significance (p < 5 × 10^−8^) or the suggestive threshold (5 × 10^−8^ ≤ p < 1 × 10^−6^) in the SNP-level analysis. For genome-wide significant loci, “nearby genes” were defined as genes whose gene bodies fell within ±250 kb of each lead variant, based on NCBI build 37.3 gene coordinates (NCBI37.3.gene.loc; GRCh37/hg19).
For suggestive loci, we deliberately took a more conservative approach to avoid overinterpreting weaker association signals. Instead of including all genes within ±250 kb, we used the “Top loci” summary from the LocusZoom web portal (LocusZoom.js; 1000 Genomes Phase 3 East Asian [EAS] LD; GRCh37) to define independent loci and then selected only the nearest gene(s) reported for each lead SNP.
Gene symbols were harmonized against the GRCh37 annotation. Symbols corresponding to known synonyms or legacy names were replaced with their current HGNC/NCBI symbols (e.g., CNT5→CNTN5 and NIBAN1→FAM129A). At the IFNG locus, the antisense transcript IFNG-AS1 was mapped to the protein-coding gene IFNG for pathway interpretation. Composite labels for intergenic regions (e.g., NPHP3–ACAD11) were split into the corresponding individual genes when both were present in the GRCh37 gene set. Entries not represented as annotated genes in GRCh37 (e.g., MYMX, LINC02391, ANKRD26P2, and ME2P1) were excluded.
The resulting non-redundant list of genes derived from genome-wide significant and suggestive loci was used as the input for the PANTHER Overrepresentation Test (PANTHER v19.0; Annotation Version Release 20240620), with the Homo sapiens genome as the reference background. Fisher’s exact test with BH-FDR correction was applied. Statistical significance was defined as FDR < 0.05, and 0.05 ≤ FDR < 0.10 was considered suggestive. Fold enrichment was defined as (list %)/(reference %). UNCLASSIFIED categories were excluded from reporting.
5. Conclusions
We conducted a case–control genome-wide association study (GWAS) in a multicenter Japanese cohort (192 cases/15,302 controls) and identified eight genome-wide significant loci (FHIT, LHX2, TRMT1L, MEGF10, SPATS1, SAMD5, MYT1L and ID4) under favorable genomic control (λ_GC = 1.04). At the gene level (MAGMA), FHIT, TRMT1L and MEGF10, together with RNF2, SWT1, VAMP1, TAPBPL and C9orf3, achieved an FDR < 0.05. Although the pathway analysis (PANTHER) did not reach FDR significance, nominal enrichments were observed in categories including inflammation mediated by chemokine and cytokine signaling, and gonadotropin-releasing hormone receptor signaling. Taken together, these findings suggest a dual-pathway working model for iSSNHL susceptibility and clinical heterogeneity, involving immune-inflammatory mechanisms and cellular stress–homeostasis; however, pathway-level interpretations are exploratory and require replication and functional validation.
Future work should focus on replication in independent cohorts, fine mapping, and functional assays in relevant cells (e.g., expression analysis, eQTL mapping, and CRISPR perturbation) to identify causal variants and molecular mechanisms. From a clinical perspective, combining genotype with acute phase biomarkers (IFN-γ/TNFα, NF-κB target gene expression, etc.) could facilitate patient stratification to predict steroid responsiveness and optimize selective anti-inflammatory therapies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chandrasekhar S.S. Do T.B.S. Schwartz S.R. Bontempo L.J. Faucett E.A. Finestone S.A. Hollingsworth D.B. Kelley D.M. Kmucha S.T. Moonis G. Clinical Practice Guideline: Sudden Hearing Loss (Update)Otolaryngol. Head Neck Surg.2019161 S 1S 4510.1177/019459981985988531369359 · doi ↗ · pubmed ↗
- 2Nakashima T. Sato H. Gyo K. Hato N. Yoshida T. Shimono M. Teranishi M. Sone M. Fukunaga Y. Kobashi G. Idiopathic sudden sensorineural hearing loss in Japan Acta Otolaryngol.20141341158116310.3109/00016489.2014.91940625315915 PMC 4266072 · doi ↗ · pubmed ↗
- 3Merchant S.N. Adams J.C. Nadol J.B.Jr. Pathology and pathophysiology of idiopathic sudden sensorineural hearing loss Otol. Neurotol.20052615116010.1097/00129492-200503000-0000415793397 · doi ↗ · pubmed ↗
- 4Li G. You D. Ma J. Li W. Li H. Sun S. The Role of Autoimmunity in the Pathogenesis of Sudden Sensorineural Hearing Loss Neural Plast.201813769147310.1155/2018/7691473 PMC 602046530008743 · doi ↗ · pubmed ↗
- 5Paprocki J. Sutkowy P. Piechocki J. Woźniak A. Markers of Oxidant–Antioxidant Equilibrium in Patients with Sudden Sensorineural Hearing Loss Treated with Hyperbaric Oxygen Therapy Oxidative Med. Cell. Longev.20196847234610.1155/2019/847234630881599 PMC 6381569 · doi ↗ · pubmed ↗
- 6Cao Z. Gao J. Huang S. Xiang H. Zhang C. Zheng B. Zhan X. Chen R. Chen B. Genetic factors associated with sudden sensorineural hearing loss: A systematic review Audiol. Neurotol.201924637410.1159/00049703230870848 · doi ↗ · pubmed ↗
- 7Kitoh R. Nishio S.-Y. Ogawa K. Okamoto M. Kitamura K. Gyo K. Sato H. Nakashima T. Fukuda S. Fukushima K. SOD 1 gene polymorphisms in sudden sensorineural hearing loss Acta Otolaryngol.201613646546910.3109/00016489.2015.111604726882452 · doi ↗ · pubmed ↗
- 8Kitoh R. Nishio S.Y. Usami S.I. Prognostic impact of gene polymorphisms in patients with idiopathic sudden sensorineural hearing loss Acta Otolaryngol.2017137 S 24S 2910.1080/00016489.2017.129697128366034 · doi ↗ · pubmed ↗