The Clinical Details of MYH9-Related Disease and DFNA17 in a Large Japanese Hearing Loss Cohort
Shinichi Goto, Akira Sasaki, Shin-ya Nishio, Chikako Shinkawa, Kiyoshi Oda, Tetsuro Wada, Kotaro Ishikawa, Tetsuo Ikezono, Shin-ichiro Oka, Nobuhiro Nishiyama, Taku Ito, Marina Kobayshi, Kozo Kumakawa, Naoko Sakuma, Hiroshi Nakanishi, Chihiro Morimoto, Natsumi Uehara

TL;DR
This study examines hearing loss caused by MYH9 gene variants in a large Japanese cohort, revealing patterns of deterioration and clinical features.
Contribution
The study provides detailed clinical insights into MYH9-related hearing loss and DFNA17 in a large patient cohort.
Findings
24 patients from 18 families were identified with MYH9-associated hearing loss.
Hearing deterioration can progress rapidly, worsening by about 50 dB within 5 years.
Patients with myosin head domain variants experience more rapid hearing loss progression.
Abstract
Background/Objectives: MYH9 gene variants cause MYH9-related disease (MYH9-RD), which is also known as Epstein syndrome, Fechtner syndrome, May–Hegglin anomaly, and Sebastian syndrome. MYH9-RD is characterized by sensorineural hearing loss, macrothrombocytopenia, thrombocytopenia, hematuria/proteinuria, glomerulonephritis, cataracts purpura, and mucosal bleeding. In addition, the MYH9 gene is also known to be causative of autosomal dominant non-syndromic hearing loss (DFNA17). MYH9-RD is a relatively rare disorder, and the detailed clinical features and mutational spectra remain unclear. Methods: In this study, we performed next-generation sequencing analysis for 15,684 hearing loss patients and identified MYH9-associated hearing loss patients. Detailed clinical information was collected for these patients and summarized. Results: In this study, we identified 24 patients from 18…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1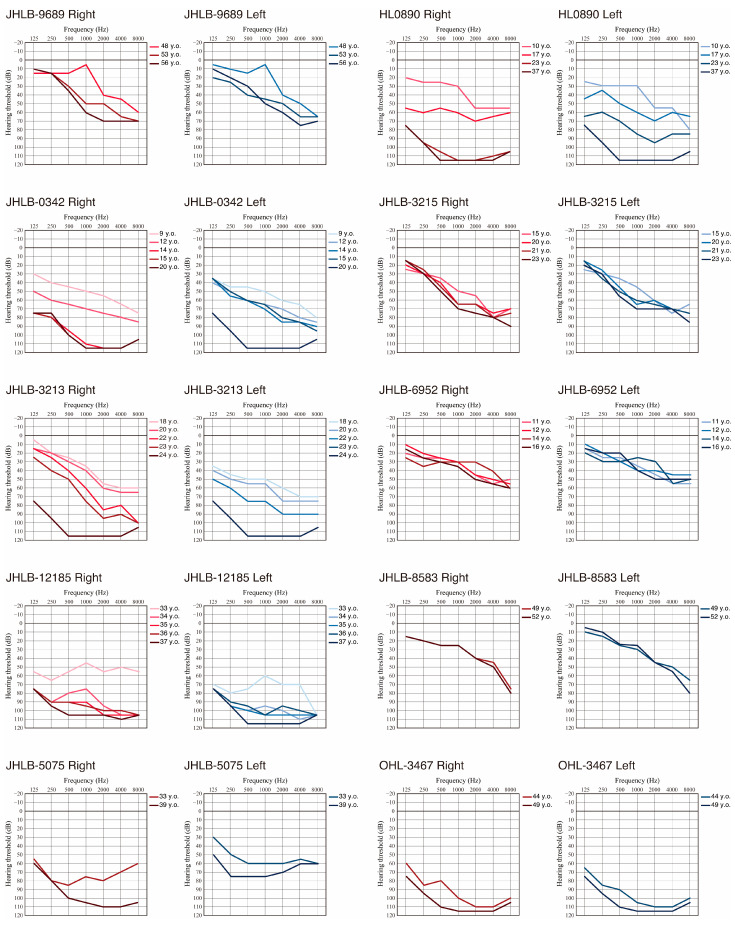 Figure 2
Figure 2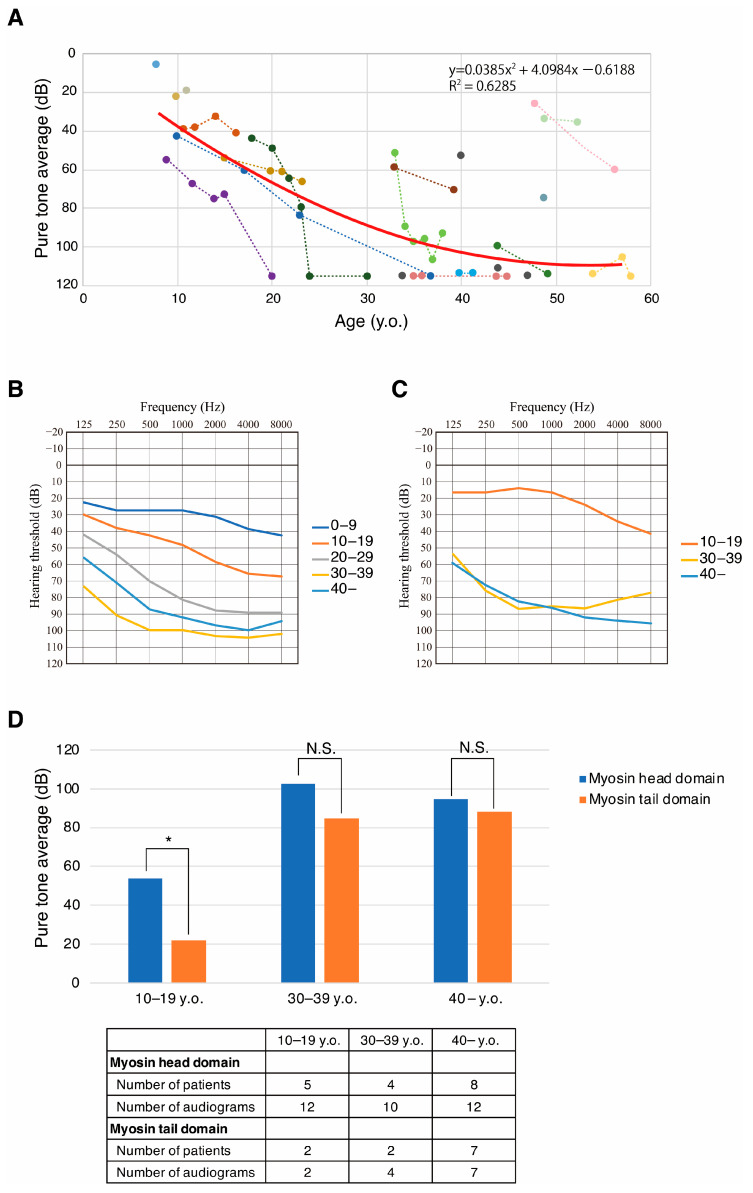 Figure 3
Figure 3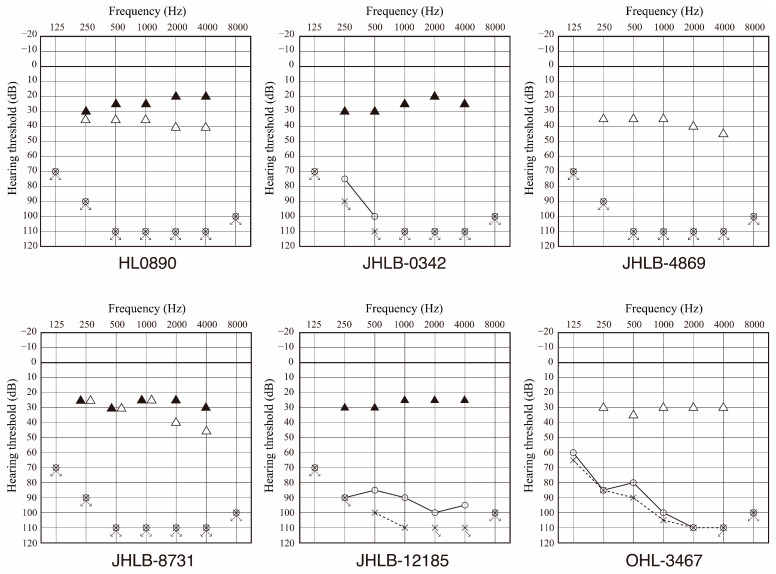 Figure 4
Figure 4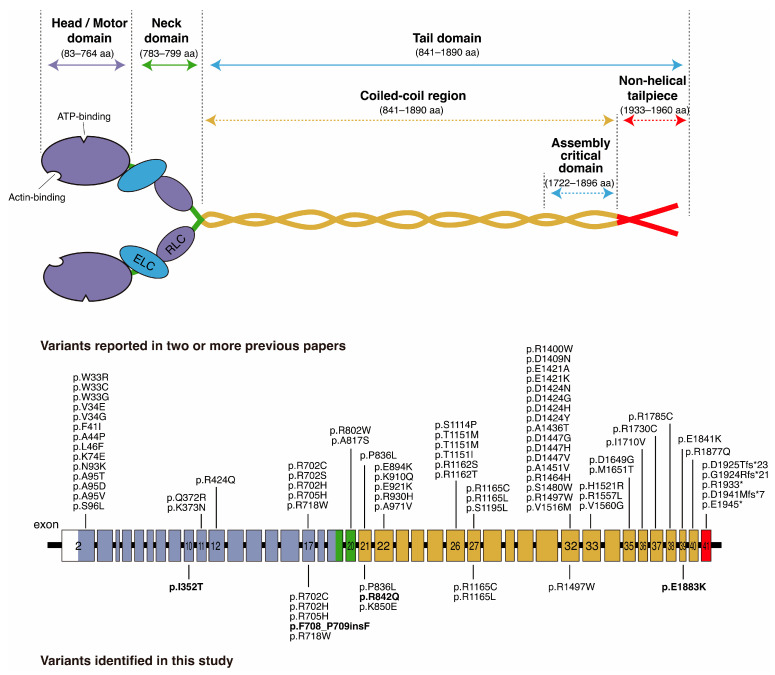 Figure 5
Figure 5- —a Health and Labor Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of J
- —Japan Agency for Medical Research and Development (AMED)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSkin and Cellular Biology Research · Hearing, Cochlea, Tinnitus, Genetics · Connective tissue disorders research
1. Introduction
Hearing loss (HL) is one of the most frequent sensory disorders, with more than 150 genes identified as causes of HL [1]. Identification of the causative gene of HL enables us to estimate the type of HL, the progressiveness of HL, and the associated symptoms. Based on the epidemiological analysis, 80% of cases of congenital HL are non-syndromic, whereas the remaining 20% are syndromic and characterized by HL with associated symptoms [2]. Most cases of syndromic HL can be diagnosed based on the combination of symptoms; however, it is sometimes difficult to diagnose patients based only on clinical phenotype, particularly in cases with late-onset symptoms or an incomplete combination of symptoms.
MYH9 gene (MIM *160775) variants are known to cause either autosomal dominant syndromic HL known as MYH9-related disease (MYH9-RD, MIM #155100) [3] or autosomal dominant non-syndromic HL (DFNA17, MIM #603622) [4]. MYH9-RD was first reported as four different syndromes: Epstein syndrome, Fechtner syndrome, May–Hegglin anomaly, and Sebastian syndrome. However, after genetic analysis, all these syndromes were found to be caused by genetic variants in the MYH9 gene and came to be regarded as a single spectrum disorder. The characteristic clinical feature of MYH9-RD is congenital macrothrombocytopenia. This macrothrombocytopenia is characterized by a triad of large platelets, thrombocytopenia, and distinctive Döhle body-like leukocyte inclusions [5]. Most cases develop late-onset sensorineural hearing loss (SNHL), protein-uric nephropathy, presenile cataract, and/or alteration of liver enzymes [3,6]. Sensorineural hearing loss (SNHL) is observed in about half of the patients, and while some patients show mild-to-moderate SNHL, others show progressive HL that finally deteriorates to severe-to-profound HL [7,8]. The patients with severe-to-profound SNHL tend to have early-onset HL [9].
The MYH9 gene is located on 22q12.3 and encodes the non-muscle myosin heavy chain IIA (NMMHC-IIA) [10,11]. The MYH9 gene contains 41 exons and consists of an N-terminal head domain and a C-terminal tail domain. NMMHC-IIA expresses most cell types and tissues, including granulocytes, megakaryocytes, and glomerular epithelial cells [12]. According to studies using rodent inner ears, NMMHC-IIA is also expressed predominantly in the hair cells of the stereocilia, spiral ligament, and spiral limbus [13].
To date, 240 MYH9 gene variants have been reported as pathogenic variants [14]. These previous studies have described mainly the clinical phenotypes of macrothrombocytopenia, with few reports focusing on SNHL. Therefore, the detailed characteristics of SNHL, such as its progressiveness or severity, remain unclear. In addition, the genotype–phenotype correlations associated with SNHL remain to be clarified. In this study, we aimed to elucidate the variant spectrum of the MYH9 gene and obtain a more precise description of the features associated with MYH9-associated HL.
2. Materials and Methods
2.1. Subjects
For this study, a total of 15,684 Japanese HL patients were enrolled from 102 otolaryngology departments nationwide, as described in our previous reports [15]. Written informed consent was obtained from all patients (or from their next of kin, caretaker, or legal guardian in case of minors or children). The Shinshu University Ethical Committee as well as the respective Ethical Committees of the other participating institutions approved this study. This study was conducted in accordance with the Declaration of Helsinki and the protocol was approved by the Ethics Committee of Shinshu University School of Medicine (No.387—4 September 2012, No.576—2 May 2017 and No.718—7 March 2022).
2.2. Clinical Evaluations
The onset age of HL, family history, the degree of progressiveness, and associated symptoms (including macrothrombocytopenia, leukocyte inclusion, hematuria, proteinuria, glomerulonephritis, cataracts, purpura, and mucosal bleeding) were analyzed based on the medical charts of the probands and their family members harboring the same MYH9 variants. Pure-tone average (PTA) was calculated from the audiometric thresholds at four frequencies (0.5, 1, 2, and 4 kHz). The severity of HL was classified into five categories: mild HL (>25 dB and ≤40 dB HL), moderate HL (>40 dB and ≤70 dB HL), severe (>70 dB and ≤90 dB HL), and profound (>90 dB HL). Asymmetric HL was defined as a difference in PTA of over 20 dB between the right and left ears. The audiometric configurations were categorized into low-frequency, mid-frequency (U-shaped), high-frequency, flat type, and deaf, as reported previously [16]. Statistical analysis was performed using Welch’s t-test. Missing data were removed from the analysis.
2.3. Next-Generation Sequencing and Bioinformatic Analysis
An amplicon library was prepared with an Ion AmpliSeq Library Kit 2.0 (ThermoFisher Scientific, Waltham, MA, USA) and Ion AmpliSeq^TM^ Custom Panel (ThermoFisher Scientific) for 158 target genes reported to cause non-syndromic HL and syndromic HL [17]. After the amplicon libraries were prepared, next-generation sequencing analysis was performed using an Ion S5 plus instrument with an Ion 540 Kit-Chef and Ion 540 Chip Kit (ThermoFisher Scientific) according to the manufacturer’s instructions. The sequence data mapping was performed against the human genome sequence (build GRCh37/hg19) using the Torrent Mapping Alignment Program. After sequencing mapping, the DNA variant regions were piled up with Torrent Variant Caller plug-in software ver. 5.16. The impacts of the variants were investigated using ANNOVAR software ver. 2020-06-08 [18]. Variants were further selected as <1% of minor allele frequencies (MAFs) in several control databases, including the 1000 genome database (http://www.1000genomes.org/), The Genome Aggregation Database (https://gnomad.broadinstitute.org), the 60,000 Japanese genome variation database ToMMo 60KJPN (https://jmorp.megabank.tohoku.ac.jp/), and the 333 in-house Japanese normal hearing controls. All filtering was carried out using our original database software, as previously reported [19]. Direct Sanger sequencing was performed to confirm the identified variants and family segregation analysis. The pathogenicity of the identified variants was evaluated in accordance with the American College of Medical Genetics (ACMG) standards and guidelines [20] and the ClinGen Hearing Loss Clinical Domain Working Group expert specification [21]. The variants classified as “Likely Pathogenic” or “Pathogenic” in the ACMG guidelines were considered to be causative variants. Furthermore, variants identified as being of “Uncertain Significance” were also considered to be causal if all of the following requirements were satisfied: (1) no other candidate variants were identified in the other 157 genes; (2) the MAF was extremely low in the control populations in gnomAD, ToMMo 60KJPN, and in-house controls; (3) the pathogenic impact was supported by the majority of the in silico prediction scores; (4) no contradictory evidence exists regarding the pathogenicity of the detected candidate variant.
3. Results
3.1. Identified Variants
As a result of the next-generation sequencing analysis for the large HL Japanese cohort, we identified 13 possibly disease-causing MYH9 variants in 18 independent families with HL. The variants identified in this study are summarized in Table 1. Among the thirteen variants, four variants were novel and nine variants were previously reported as causative for MYH9-associated HL. Six variants were located in the myosin head domain (HD) and four were located in the myosin tail domain (TD). All novel variants identified in this study were missense or non-frameshift insertion variants, and these variants were classified as variants of “Uncertain Significance” based on the ACMG guidelines. However, we regarded these variants as candidate causative variants for MYH9-associated HL as they fulfilled the criteria described in the Methods Section. Most of the in silico pathogenicity prediction scores supported the pathogenicity of the identified variants. In addition, MAFs in the control populations fulfilled the criteria supporting their pathogenicity defined by the ClinGen HL expert panel (MAF ≤ 0.00002). However, all novel variants were of “Uncertain Significance,” and further studies are needed to confirm the pathogenicity of those variants.
3.2. Characteristics of MYH9-Associated Hearing Loss and Outcomes of Cochlear Implantation
The pedigrees and audiograms of the MYH9-associated HL patients identified in this study are shown in Figure 1, and clinical characteristics are summarized in Table 2. In this study, we identified 24 patients with MYH9-associated HL from 18 independent families. A total of ten patients were male, and fourteen patients were female. Among the 18 families, 13 had an autosomal dominant (AD) family history, while 4 were sporadic cases without any affected family member. Regarding the onset age of the HL, 4 patients had prelingual onset HL (below the age of 6) and 20 patients had post-lingual onset HL. The post-lingual onset age ranged widely from 6 to 45 years. The hearing levels at the examination varied from mild to profound. Ten patients had profound HL, three patients had severe HL, and nine patients had mild-to-moderate HL. Only two patients under 10 years of age (son of JHLB-9870 and grandson of JHLB-3212) have normal hearing while harboring the MYH9 variant, although other family members with the same variant showed moderate-to-profound HL. Most of the patients with mild-to-moderate HL showed high-frequency sloping-type HL. We found that 91% (20/22) of patients were aware of their hearing loss progression and almost half of the patients had tinnitus (56.5%, 13/23). On the other hand, only 13% (3/23) of patients had a history of vertigo.
To clarify the hearing deterioration of MYH9-associated HL, we collected serial audiograms for 10 patients. As shown in Figure 2, eight cases, excluding JHLB-6952 and JHLB8583, showed hearing deterioration within the observation period. It is noteworthy that several cases showed asymmetrical hearing deterioration (see the 17 and 23 y.o. hearing thresholds for JHLB-0890, the 14 and 15 y.o. hearing thresholds for JHLB-0342, the 18 -23 y.o. hearing thresholds for JHLB-3213, the 33 and 34 y.o. hearing thresholds for JHLB-12185, and the 33 and 39 y.o. hearing thresholds for JHLB-5075 in Figure 2), finally deteriorating to bilateral profound HL. In addition, HL0890, JHLB-0342, JHLB-3213, and JHLB12185 showed rapid hearing deterioration, which worsened by about 50 dB (ranging from 48 dB to 67 dB) within five years. The detailed mechanism underlying this rapid hearing deterioration remains unclear, but careful follow-up is necessary for MYH9-associated HL patients due to the potential for rapid hearing deterioration.
Scatter plotting of hearing thresholds and audiometric testing age is shown in Figure 3A. As shown in Figure 3A, the hearing threshold for most patients showed hearing deterioration with age, with most cases showing profound HL after 40 years of age. In addition, we compared mean hearing thresholds every 10 years for the patients carrying HD variants and those carrying TD variants (Figure 3B,C). As shown in Figure 3B,C, MYH9-associated HL patients carrying HD variants showed a stronger tendency for rapid hearing deterioration compared to those carrying TD variants. We also compared the PTA for the patients carrying HD variants and those carrying TD variants (Figure 3D), and found that patients with HD variants had more statistically severe HL than patients with TD variants in the 10–19 y.o. age group (p = 0.00027, Welch’s t-test), but not in the 30–39 y.o. or ≥40 y.o. age groups. However, the number of patients in the 10–19 y.o. age group was small and further investigation is still needed.
Nine patients received cochlear implantation (CI). We obtained hearing thresholds after CI for six patients and found that the averaged hearing threshold was 30.5 dB (range 22.5 to 38.8 dB) (Figure 4). Average pre-CI hearing level in PTA for the same six patients was 108.1 dB (range 95 to 110 dB), and CI improved hearing level significantly (p = 2.3 × 10^−13^, Welch’s t-test). In addition, monosyllable perception score in a quiet setting was 75% (range 60 to 100%) on average. Thus, CI is a good treatment option for the patients with MYH9-associated HL.
3.3. Associated Symptoms of MYH9-Associated HL Patients Identified in This Study
The clinical features are shown in Table 2. We obtained clinical histories for thrombocytopenia from 18 patients, and found that 7 patients had thrombocytopenia (7/18; 38.9%) and 8 patients had macrothrombocytopenia (8/13; 61.5%). We obtained data regarding leukocyte inclusion from only four patients, and found that only one patient had leukocyte inclusion (1/4; 25%). Four patients had purpura (4/15; 26.7%) and four patients had mucosal bleeding (4/14; 28.6%).
We obtained clinical histories for hematuria or proteinuria from 13 patients, and found that 4 patients had hematuria/proteinuria (4/13; 30.8%). Furthermore, three patients had glomerulonephritis (3/12; 25.0%) and two of the three patients had hematuria/proteinuria. We also obtained clinical histories for cataracts from 14 patients, and found that only 1 patient had cataracts (1/14; 7.1%).
4. Discussion
The MYH9 gene encodes myosin heavy chain 9 non-muscle, which is known as non-muscle myosin-IIA [10,11]. The pathogenic variants in the MYH9 gene cause MYH9-RD or DFNA17 [3,4]. In this study, we identified 13 variants in 24 HL patients from 18 different families. Thus, the prevalence of MYH9-associated HL in this study was 0.11% (18/15,684) in Japanese HL patients.
Among the 13 variants, 6 were HD and 6 were TD variants (Figure 5). It has been reported that patients with pathogenic variants in the HD have a higher risk of early-onset and severe deafness than patients with variants in the TD [7,28,29]. Pecci et al. reported that amino acid substitutions of arginine 702 of the HD are related to the most severe phenotype: all individuals with mutations involving this residue experience end-stage renal disease and profound HL before the fourth decade [28]. On the other hand, p.Asp1424His variants in the coiled-coil region are associated with late-onset symptoms. This variant disrupts the interactions between the SH3 motif and the 50 kDa sub-domain of the HD and increases the risk of deafness while decreasing the risk of kidney disorders or cataracts. Variants involving the arginine 1165 residue in the coiled-coil region also showed a similar phenotype [30]. In this study, patients with variants in either the HD or the TD demonstrated progressive HL (Figure 3B,C). Similarly to previous reports, our patients with variants in the HD showed a tendency for more severe HL compared with the patients with TD variants (Figure 3B–D). However, the number of patients in each age group used in this study was relatively small and further investigation is still needed.
Pecci et al. reported that the HL types in MYH9-associated HL patients were mostly sloping-type [8]. In the cases with DFNA17, a mild-to-moderate sloping-type HL is first observed, followed by deterioration of the low-frequency hearing level and eventual progression to severe, flat-type HL [32]. We observed a similar progression in hearing loss to that reported in previous studies (Figure 2).
Nine patients received CI with favorable outcomes. In previous studies, several patients with MYH9-RD underwent CI and almost all of the patients had effective outcomes with better postoperative speech perception scores after CI [8,28,33,34,35]. According to studies on the inner ear of rodents, NMMHC-IIA is expressed in the hair cells of the stereocilia, spiral ligament, and spiral limbus [13]. Good CI performance can be expected because MYH9 expression is limited to the hair cells of the stereocilia, spiral ligament, and spiral limbus, and the spiral ganglions appear to remain intact. In addition, it is important to manage bleeding during CI surgery for patients with thrombocytopenia. In our study, we obtained information regarding CI surgery in one patient, and found that the surgery was performed without major bleeding complications. On the other hand, there have been reports of cases in which platelet transfusion was performed preoperatively to prevent perioperative bleeding [34,35]. Mori et al. reported a case where eltrombopag, a nonpeptide agonist of the thrombopoietin receptor, was used, meaning that they could avoid using platelet transfusion [33]. Therefore, CI is a safe procedure in MYH9-RD patients whenever adequate preventative measures are taken.
In our study, the prevalence of thrombocytopenia was low (38.9%: 7/18) compared to that in previous studies [8,28,36,37]. Similarly, the prevalence of mucosal bleeding was also low (28.6%: 4/14). Eight patients were observed to have macrothrombocytopenia (61.5%: 8/13). In the case of macrothrombocytopenia, automated cell counters usually underestimate platelet counts because platelets are mainly identified based on size [38]. It is important to avoid unnecessary blood transfusions when performing CI surgery in such cases. In previous studies, all patients with MYH9 variants had macrothrombocytopenia, which differs from the findings in this study [8,28]. Leukocyte inclusions involving Döhle-like bodies were present in 42–84% of MYH9-RD patients [3,39,40]. However, there was only one patient with leukocyte inclusion in our study. It is noteworthy that there were a variety of clinical phenotypes associated with thrombocytopenia or macrothrombocytopenia, even in the patients carrying the same variant (e.g., patients carrying the c.2114G > A variant). In a previous study, the c.2114G > A variant was also identified from non-syndromic hearing loss (DFNA17) patients [31]. On the other hand, the prevalences of hematuria/proteinuria (30.8%: 4/13), glomerulonephritis (25.0%: 3/12), and cataracts (7.5%: 1/14) were comparable to those in previous reports [8,28].
In this study, we mainly analyzed non-syndromic HL patients from collaborating otolaryngology departments nationwide, so we could access not only MYH9-RD but also DFNA11 patients who did not show any associated symptoms. However, to avoid the possibility of overlooking associated symptoms, we need to collaborate with other departments, such as hematology and nephrology departments, in future studies.
5. Conclusions
In this study, we were able to clarify the detailed characteristics of HL associated with MYH9-RD and DFNA17 in a relatively large number of patients. One of the remarkable results of this study was our ability to clarify the details of hearing deterioration using serial audiogram data collected from the same patients. Similarly to previous reports, patients with HD variants showed a tendency for relatively severe HL in comparison to those with TD variants, but both cases deteriorated to profound HL. In addition, we showed that CI was an effective treatment option for patients with severe-to-profound HL. The results identified in this study will be beneficial in enabling more appropriate treatment for patients with MYH9-associated HL based on the expectation of future hearing deterioration and favorable CI outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Walls W.D. Azaiez H. Smith R.J.H. Hereditary Hearing Loss Homepage Available online: https://hereditaryhearingloss.org(accessed on 15 November 2025)
- 2Ahmed S. Sheraz S. Malik S.A. Ahmed N.R. Malik S.A. Farooq S. Raheem A. Basheer F. Nayyar Z.A. Malik F.E. Frequency of Congenital Hearing Loss in Neonates J. Ayub. Med. Coll. Abbottabad 20183023423629938425 · pubmed ↗
- 3Seri M. Pecci A. Di Bari F. Cusano R. Savino M. Panza E. Nigro A. Noris P. Gangarossa S. Rocca B. MYH 9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness Medicine 20038220321510.1097/01.md.0000076006.64510.5c 12792306 · doi ↗ · pubmed ↗
- 4Lalwani A.K. Goldstein J.A. Kelley M.J. Luxford W. Castelein C.M. Mhatre A.N. Human nonsyndromic hereditary deafness DFNA 17 is due to a mutation in nonmuscle myosin MYH 9Am. J. Hum. Genet.2000671121112810.1016/S 0002-9297(07)62942-511023810 PMC 1288554 · doi ↗ · pubmed ↗
- 5Kunishima S. Kojima T. Matsushita T. Tanaka T. Tsurusawa M. Furukawa Y. Nakamura Y. Okamura T. Amemiya N. Nakayama T. Mutations in the NMMHC-A gene cause autosomal dominant macrothrombocytopenia with leukocyte inclusions (May-Hegglin anomaly/Sebastian syndrome)Blood 200197114711491115955210.1182/blood.v 97.4.1147 · doi ↗ · pubmed ↗
- 6Pecci A. Biino G. Fierro T. Bozzi V. Mezzasoma A. Noris P. Ramenghi U. Loffredo G. Fabris F. Momi S. Alteration of liver enzymes is a feature of the MYH 9-related disease syndrome P Lo S ONE 20127 e 3598610.1371/journal.pone.003598622558294 PMC 3338476 · doi ↗ · pubmed ↗
- 7De Rocco D. Zieger B. Platokouki H. Heller P.G. Pastore A. Bottega R. Noris P. Barozzi S. Glembotsky A.C. Pergantou H. MYH 9-related disease: Five novel mutations expanding the spectrum of causative mutations and confirming genotype/phenotype correlations Eur. J. Med. Genet.20135671210.1016/j.ejmg.2012.10.00923123319 PMC 3546164 · doi ↗ · pubmed ↗
- 8Pecci A. Verver E.J. Schlegel N. Canzi P. Boccio C.M. Platokouki H. Krause E. Benazzo M. Topsakal V. Greinacher A. Cochlear implantation is safe and effective in patients with MYH 9-related disease Orphanet J. Rare Dis.2014910010.1186/1750-1172-9-10024980457 PMC 4105151 · doi ↗ · pubmed ↗