Mesenchymal Stromal Cells and Extracellular Vesicles: A Novel Therapeutic Paradigm for Mitochondrial Dysfunctions
Eman Salem Algariri, Fazlina Nordin, Min Hwei Ng, Izyan Mohd Idris, Norwahidah Abdul Karim, Gee Jun Tye, Wan Safwani Wan Kamarul Zaman

TL;DR
This paper reviews how mesenchymal stromal cells and their extracellular vesicles may offer new treatments for mitochondrial dysfunction by restoring cellular energy and reducing stress.
Contribution
The paper introduces MSCs and MSC-EVs as a novel therapeutic approach for mitochondrial disorders.
Findings
MSCs and MSC-EVs can enhance mitochondrial respiration and cellular bioenergetics.
They reduce oxidative stress and transfer functional mitochondrial components.
These therapies show promise across various disease models.
Abstract
Mitochondrial dysfunction is a central pathological feature of a wide range of inherited and acquired disorders and is characterized by impaired oxidative phosphorylation, disrupted cellular energy metabolism, and excessive oxidative stress. Although advances in molecular diagnostics have improved disease recognition, effective disease-modifying therapies remain limited, and clinical outcomes are often suboptimal, highlighting the need for novel therapeutic strategies. Mesenchymal stromal cells (MSCs) and their extracellular vesicles (MSC-EVs) have emerged as promising candidates for targeting mitochondrial dysfunction due to their regenerative, immunomodulatory, and metabolic regulatory properties. In this review, we provide a comprehensive overview of recent in vitro and in vivo studies investigating the capacity of MSCs and MSC-EVs to restore mitochondrial function by enhancing…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
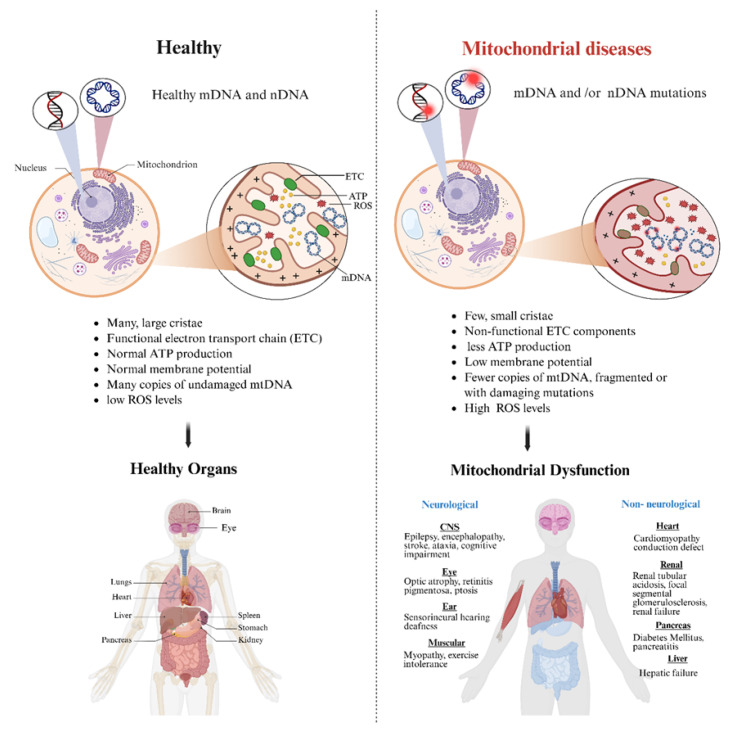 Figure 1
Figure 1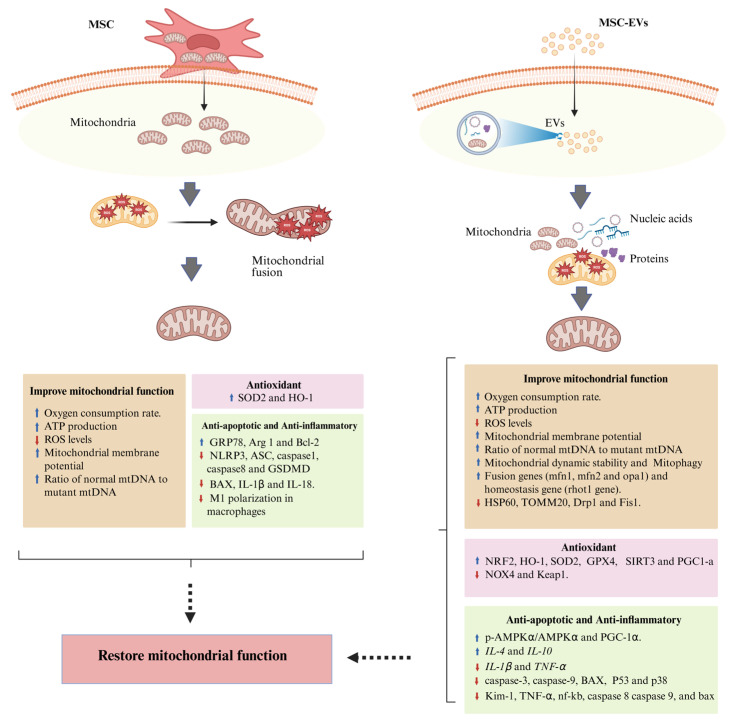 Figure 2
Figure 2| Source of MSCs/MSC-EVs | Targeted Disease | Targeted Cells/Animal Model | Dosage/Route of Administration | Therapeutic Effects | Mechanism | Ref. |
|---|---|---|---|---|---|---|
| Human BM-MSCs & highly purified MSCs (RECs) | MELAS syndrome | iPSC-derived MELAS neurons | Not specified | - Restore mitochondrial membrane potential | - Mitochondria donation | [ |
| Human MSCs | Leber’s hereditary optic neuropathy (LHON) | LHON iPSC-derived NPCs | 1:1 ratio MSC: NPCs | - Increase mitochondrial respiration and ATP production | - Mitochondria donation | [ |
| Human BM-MSCs | Complex I deficiency | Human fibroblast with MT-ND3 & MT-ND6 | Not specified | - Improve mitochondrial respiration | - Mitochondria donation | [ |
| Human UC-MSCs | Diabetic nephropathy | Murine macrophage cell line (RAW264.7 cells) | 1:2 ratio MSCs: RAW264.7 cells | - Anti-inflammatory effect | - Mitochondria donation | [ |
| 8-week-old male CD1 mouse model of diabetic nephropathy | Mice were injected intravenously with 5.0 × 105 UC-MSCs thrice every 4 weeks | |||||
| Human and murine BM-MSCs | Diabetic nephropathy | Human podocytes | 1:1 ratio MSCs: podocytes | - Improve mitochondrial function | - Mitochondria donation | [ |
| 8-week-old male C57BL6 mouse model of diabetic nephropathy | Mice were injected via tail vein, with BM-MSCs (1.0 × 104 cells/g body weight) once a week for 6 consecutive weeks | |||||
| Human OM- | Cerebral ischemia/reperfusion injury | Neuron (SH-SY5Y) cells | Not specified | - Improve neuron mitochondrial function (increase MMP and decrease ROS) | - Increase GRP78 and Bcl-2 proteins | [ |
| Adult Sprague–Dawley rat | IhOM-MSCs (1 × 106) were injected into the rat tail vein | |||||
| Mouse Ad-MSC-EVs | Leber’s hereditary optic neuropathy (LHON) | LHON model cells (GM10742 cells) | 6 μg EV-Mito protein/well | - Restore mitochondrial functions (MMP, ATP production, mitochondrial ROS levels, and mPTP opening) | - Mitochondria donation | [ |
| 3-month-old mutant mtND4R340H mtTg LHON male mice | Mice received an intravitreal injection of 1 μg EV-Mito protein/eye, 2 times administrations | |||||
| Human UC-MSC-EV | Cardiac hypertrophy | Neonatal rat cardiac myocytes (from 1- to 3-day-old Sprague–Dawley rats) as model of cardiac hypertrophy | - 100 ug/mL Nor-EVs or Hypo-EVs | - Reduce the cardiomyocyte size | - Transfer DJ-1 protein to cardiomyocytes | [ |
| Male 8-week-old adult C57BL/6 mice as model of cardiac hypertrophy | 200 μg/100 μL of Nor-EVs or Hy-EVs were injected into mice through caudal vein once a week, from one week after surgery for 3 weeks | |||||
| Human UC-MSC-Exos | Premature ovarian insufficiency | KGN cells as model of POI | Nor-Exos and hy-Exos (50 µg/mL) | - Enhance mitochondrial function and regulate mitochondrial oxidative stress | - Increase expression of | [ |
| 8-week-old SD female rats’ model of POI | 200 µL of Hy-Exos or Nor-Exos (1 × 109 cells) was transplanted into each ovary for two weeks (once a week) | |||||
| Mouse MSC /MSC-Exo | Cigarette smoking induced mitochondrial dysfunction | lung epithelial cells (BEAS2B cells) exposure to cigarette smoke (CS) | Not specified | - Protective response against the CSE-altered mitochondrial respiration | - Increase the expression of fusion genes (mfn1, mfn2 and opa1) and mitochondrial homeostasis gene (rhot1 gene) | [ |
| Mouse HF-MSC-Exos | Ulcerative colitis (with high mitochondrial fission/fusion) | LPS-treated mouse MODE-K cells as a model of ulcerative colitis | Not specified | - Alleviate mitochondrial dysfunction and oxidative stress | - Reduce the expression of HSP60, TOMM20, Drp1 and Fis1 | [ |
| 4–6 weeks C57BL/6J mice | 100 μg of Nor-Exos and Hy-Exos were injected via the tail vein | |||||
| Human Ad-MSC-Exos | ALI | MH-S mouse macrophage cells | Exosomes (10 μg/mL) | - Improve macrophages’ mitochondrial integrity and oxidative phosphorylation level | - Transfer mitochondrial components | [ |
| Human UC-MSC-Exos | IVDD | Human degenerative | UCMSC-exos (1011 particles/mL) | - Improve viability of NPCs | - Reduce ROS and mitochondrial superoxide levels | [ |
| 23-month-old male Sprague–Dawley rats as IVDD model (by IVD puncture) | 10 uL of UCMSC-Exos (1011 particles/mL) were injected into the punctured discs every 2 weeks for 2 months | |||||
| Human P-MSC-EVs | CKD | HK-2 as induced model of CKD | Norm-EVs or Hypo-EVs (100 μg/mL) | - Reduce renal fibrosis | - Reduce collagen I and α-SMA | [ |
| Male C57BL/6 SPF mice (7–9 weeks old) were used as a model of ischemia–reperfusion (I/R)-induced renal fibrosis | 100 μg of Norm-EVs or Hypo-EVs in 0.15 mL of PBS solvent was administered by tail vein injection immediately after the surgery and on day (D) 1 postsurgery in the Norm-EVs and Hypo-EVs groups, respectively | |||||
| Human P-MSC-EVs | Acute kidney injury | TEC line (HK-2) as model of oxidative stress | 40–120 μg P-MSC-EVs | - Restore renal function | - Modulate inflammation and inhibit apoptosis (downregulation of Kim-1, TNF-α, nf-kb, caspase 8, caspase 9, and bax) | [ |
| 6–8-week-old male FVB mice as a model of ischemia/reperfusion-induced AKI | 0.1 mL of 80 μg of EVs was injected intravenously into AKI mice |
- —Faculty of Medicine, Universiti Kebangsaan Malaysia (UKM)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsExtracellular vesicles in disease · Mitochondrial Function and Pathology · GDF15 and Related Biomarkers
1. Introduction
Mitochondria are double-membrane organelles that function as central regulators of cellular metabolism, bioenergetics, and signaling. Structurally, they consist of an outer mitochondrial membrane that mediates metabolite exchange and protein import and an inner mitochondrial membrane folded into cristae, which contain the electron transport chain (ETC) complexes I–V and ATP synthase. The inner membrane encloses the mitochondrial matrix, which harbors mitochondrial DNA (mtDNA), ribosomes, and enzymes of the tricarboxylic acid (TCA) cycle and fatty acid β-oxidation [1]. Functionally, mitochondria generate ATP via oxidative phosphorylation (OXPHOS) by coupling electron transfer to proton gradient formation and ATP synthesis. Beyond energy production, mitochondria regulate reactive oxygen species (ROS) signaling, calcium homeostasis, apoptosis through cytochrome c release, innate immune responses, and cell fate decisions [2]. Mitochondrial dynamics—including fission, fusion, mitophagy, and biogenesis—are critical for maintaining mitochondrial quality control and adapting mitochondrial function to cellular metabolic demands [3].
Consequently, defects in mitochondrial structure, bioenergetic pathways, signaling functions, or dynamic processes can lead to mitochondrial dysfunctions (MDs), ultimately disrupting cellular homeostasis and contributing to the pathogenesis of a wide spectrum of metabolic, neurodegenerative, cardiovascular, and genetic disorders. Mitochondrial dysfunctions are broadly classified into two categories: primary mitochondrial dysfunctions/diseases (PMDs), which are groups of genetic disorders caused by inherited mutations in nuclear or mitochondrial genes that encode structural OXPHOS proteins or proteins required for OXPHOS function, and secondary mitochondrial dysfunctions (SMDs), which arise as a downstream consequence of non-mitochondrial genetic defects, environmental factors, aging, and chronic or inflammatory diseases [4,5]. The hallmarks of mitochondrial dysfunctions are energy deficits, increased oxidative stress, loss of mitochondrial membrane potential, and activation of apoptotic pathways (Figure 1). PMDs are characterized by complex genetics, multisystemic involvement, and significant diagnostic challenges; a single syndrome may result from mutations in multiple genes, while a single variant may lead to widely variable clinical presentations [6]. PMDs affect approximately 1 in 5000 adults worldwide, with asymptomatic carriers reaching nearly 1 in 250 in the general population [7,8]. Due to their multisystemic impact, progressive course, and absence of curative therapies, MDs impose a substantial disease burden and highlight the urgent need for strategies capable of restoring mitochondrial function and improving organ performance.
Mesenchymal stromal cells (MSCs) have emerged as promising candidates in regenerative medicine due to their immunomodulatory properties, paracrine activity, and ability to transfer healthy organelles and bioactive molecules to damaged cells [9]. Notably, accumulating evidence indicates that many of the therapeutic benefits of MSCs are mediated through their secretome, particularly extracellular vesicles (EVs), rather than direct cell engraftment [10]. MSC-derived EVs (MSC-EVs), including exosomes, encapsulate a diverse cargo of proteins, lipids, mRNAs, microRNAs, and, in some cases, mitochondrial components or whole mitochondria, enabling them to modulate inflammation, oxidative stress, apoptosis, and cellular metabolism [11]. Both MSCs and MSC-EVs are emerging as attractive therapeutic platforms for mitochondrial restoration due to their ability to transfer bioactive molecules and mitochondrial components to injured cells. While MSCs can directly enhance tissue repair and mitochondrial function, growing evidence suggests that MSC-EVs mediate many of these effects and represent a safer, cell-free alternative with significant potential for rescuing mitochondrial dysfunction [11,12].
Therefore, this review aims to evaluate emerging therapeutic strategies based on mesenchymal stromal cells (MSCs) and their extracellular vesicles (MSC-EVs) for the treatment of mitochondrial dysfunctions. This review synthesizes current experimental and preclinical evidence regarding their capacity to restore mitochondrial function, enhance cellular bioenergetics, and attenuate oxidative stress and inflammation. Furthermore, it examines the underlying molecular mechanisms, discusses translational potential, and highlights key limitations that must be addressed for clinical application.
2. Genetics of Mitochondrial Dysfunctions
Mitochondrial DNA (mtDNA) is a small, circular, double-stranded genome located inside mitochondria, the organelles responsible for producing cellular energy through oxidative phosphorylation (OXPHOS). Unlike nuclear DNA, mtDNA is inherited almost exclusively from the mother and encodes 37 genes, including 13 proteins essential for the mitochondrial respiratory chain, 22 transfer RNAs, and 2 ribosomal RNAs [13,14]. The nuclear genome encodes approximately 250–300 proteins that localize to the mitochondria. These proteins are synthesized in the cytoplasm and transported into the organelle through an intricate protein import system. In total, an estimated 1500 nuclear genes are involved in mitochondrial processes, including functions beyond the electron transport chain (ETC) [15,16,17]. mtDNA has a high mutation rate due to limited DNA repair mechanisms and its proximity to reactive oxygen species, with the mitochondrial genome exhibiting a mutation rate approximately 100–1000 times higher than that of the nuclear genome, making it a major contributor to mitochondrial dysfunction [18,19]. Given the dual genetic control of mitochondrial function, pathogenic variants in mtDNA and/or nDNA can impair energy production and cause a wide range of multisystemic disorders affecting high-energy-demand tissues, such as the brain, heart, and skeletal muscle [4,5,20].
Primary mitochondrial diseases are caused by genetic mutations involved in mtDNA or nDNA genes that encode OXPHOS structure and function proteins [4]. Pathogenic variants can be inherited through autosomal recessive, autosomal dominant, maternal, or X-linked patterns [15]. PMD manifests in a wide range of clinical syndromes. Among the most frequently reported are mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like episodes (MELAS), Leber hereditary optic neuropathy (LHON), Kearns–Sayre syndrome (KSS syndrome), progressive extraocular muscle paralysis (PEO), and Leigh syndrome [7]. PMDs have a complex genetic basis, in which specific syndromes may arise from diverse genetic etiologies. For example, Leigh syndrome can result from a variety of mtDNA and nDNA mutations, including pathogenic variants in MT-ND1, MT-ND3, MT-ND4, and MT-ND6; nuclear defects such as NDUFS1; or a combination of both [4]. Conversely, a single mtDNA mutation may lead to multiple clinical presentations. A well-recognized example is the m.3243A>G variant in MT-TL1, which can manifest as PEO, MELAS, or maternally inherited diabetes and deafness (MIDD) [5,18].
In addition, the severity of mitochondrial diseases is strongly influenced by the degree of heteroplasmy and the tissues affected. Mutation m.8993 T>G or m.8993 T>C in the MT-ATP6 gene is implicated in the pathogenesis of neuropathy, ataxia, and retinitis pigmentosa (NARP). These mutations are also associated with Leigh syndrome, with the specific phenotype determined by the level of heteroplasmy. Levels exceeding 85% are predominantly linked to childhood-onset Leigh syndrome, whereas a level of 60–70% typically results in adult-onset NARP. Both diseases may occur with heteroplasmy levels between 70 and 85% [21,22]. Similarly, the m.13094T>C variant shows that high mutation loads are associated with severe Leigh syndrome, while lower heteroplasmy produces a broader spectrum of neurological symptoms [23].
Secondary mitochondrial dysfunction (SMD) refers to mitochondrial impairment that occurs as a downstream consequence of non-mitochondrial genetic defects, systemic diseases, environmental stressors, or aging, rather than primary mutations in oxidative phosphorylation (OXPHOS) genes. In SMD, the primary pathological insult indirectly disrupts mitochondrial bioenergetics, redox homeostasis, and quality control pathways, thereby contributing to disease progression and tissue degeneration [24,25,26]. SMD is frequently implicated in multifactorial disorders such as diabetes, cardiovascular disease, cancer, kidney disease, and neurodegenerative conditions [24,27,28,29,30]. Genetic alterations in SMD frequently involve nuclear genes that regulate mitochondrial dynamics, quality control, and metabolic pathways rather than core respiratory chain subunits. Mutations in genes controlling mitochondrial fusion and fission, such as MFN2, OPA1, and DNM1L, result in fragmented or dysfunctional mitochondrial networks that compromise ATP production and intracellular mitochondrial distribution [31,32]. Similarly, defects in mitophagy-related genes, including PINK1 and PRKN, lead to impaired clearance of damaged mitochondria, promoting oxidative stress and cellular degeneration, particularly in neurodegenerative disorders [33,34].
Alterations in genes involved in mitochondrial iron–sulfur cluster biogenesis, such as FXN, further contribute to secondary respiratory chain dysfunction and oxidative stress, as observed in Friedreich’s ataxia [35]. Moreover, mutations in genes responsible for mtDNA maintenance, including POLG, TWNK, and MPV17, can cause mtDNA depletion or deletions, resulting in secondary OXPHOS defects [36]. At the signaling level, SMD is strongly influenced by dysregulated metabolic and stress response pathways. The PGC-1α signaling axis, which controls mitochondrial biogenesis and oxidative metabolism, is frequently suppressed in metabolic and neurodegenerative diseases, leading to reduced mitochondrial mass and respiratory capacity [37,38]. Energy-sensing pathways such as AMPK and nutrient-sensing pathways such as mTOR modulate mitochondrial biogenesis, autophagy, and mitophagy, and their chronic dysregulation contributes to mitochondrial dysfunction in diabetes, aging, and cancer [39,40]. Inflammatory signaling pathways, including NF-κB and the NLRP3 inflammasome, are activated by mitochondrial ROS and mtDNA release, further exacerbating mitochondrial damage and chronic inflammation [41]. Table 1 summarizes the key genetic basis, pathophysiological mechanisms, and key clinical features of selected examples of primary and secondary mitochondrial dysfunctions.
3. Diagnosis of Mitochondrial Dysfunctions
Mitochondrial diseases are genetically complex and can present with a wide spectrum of clinical manifestations, making diagnosis challenging. Their presentation may be neurological or non-neurological, with onset ranging from acute or subacute to slowly progressive. Neurological features include seizures, ataxia, stroke-like episodes, encephalopathy, myopathy, muscle weakness, optic atrophy, retinitis pigmentosa, and auditory neuropathy. Non-neurological manifestations reflect multisystem involvement and may include hypertrophic or dilated cardiomyopathy, Fanconi syndrome, diabetes mellitus, and premature ovarian failure [52,53,54,55]. Childhood mitochondrial diseases are often associated with diverse clinical presentations, particularly in children under two years of age [56]. Common features include fatigue, vomiting, failure to thrive, hypotonia, encephalopathy, and seizures [57]. Some mitochondrial disorders follow a characteristic clinical course, such as Pearson syndrome (PS), which initially presents with severe hypoproliferative anemia in early infancy and later progresses to multi-organ dysfunction, including lactic acidosis, pancreatic insufficiency, renal tubulopathy, failure to thrive, muscle hypotonia, and endocrine abnormalities [21,22]. During disease progression, anemia may spontaneously resolve in some PS patients, whereas others may evolve into Leigh syndrome or Kearns–Sayre syndrome [22]. Because mitochondrial diseases can mimic many common conditions due to their multisystem involvement, they are frequently considered in the differential diagnosis of diverse disorders. Therefore, confirmatory analyses—including biochemical testing and molecular genetic evaluation—are essential for achieving an accurate diagnosis.
A variety of biochemical screening tests can support the diagnosis of mitochondrial diseases. These include complete blood counts, urine organic and amino acid analyses, hormone screening, hemoglobin A1C, comprehensive metabolic panels, measurements of blood lactate and pyruvate, and assessments of creatine kinase, ammonia, carnitine, acylcarnitine, and lipoprotein profiles [53]. Molecular genetic testing is a critical component of the diagnostic process, as it enables identification of the underlying molecular etiology of mitochondrial dysfunction and guides therapeutic decision-making. The first-line molecular diagnostic test for mitochondrial disease typically involves next-generation sequencing (NGS) of mitochondrial DNA (mtDNA) to detect point mutations, deletions, and heteroplasmic variants associated with primary mitochondrial diseases [58,59,60]. If mtDNA sequencing does not identify a causative mutation, whole-exome sequencing (WES) or whole-genome sequencing (WGS) can be used to detect pathogenic variants in nuclear genes encoding mitochondrial proteins [58,59]. In addition to genetic analysis, tissue-based investigations—such as biochemical assays of respiratory chain enzyme activity and histopathological evaluation of skeletal muscle or skin biopsies—can provide supportive or confirmatory evidence of mitochondrial dysfunction, particularly when molecular findings are inconclusive [58,60].
4. Treatment and Prognosis of Mitochondrial Dysfunctions
The treatment of mitochondrial diseases remains challenging due to their genetic heterogeneity, multisystem involvement, and the limited availability of disease-modifying therapies. Currently, most therapeutic approaches are supportive and aim to improve mitochondrial bioenergetics, reduce oxidative stress, and manage organ-specific complications. Conventional management strategies include metabolic supplementation and symptomatic treatment. Nutritional and pharmacological agents such as coenzyme Q10 (CoQ10), riboflavin, L-carnitine, thiamine, and antioxidant vitamins are commonly used to enhance electron transport chain (ETC) activity, improve fatty acid oxidation, and mitigate oxidative stress [5,61,62,63,64,65,66,67,68,69,70]. In clinical practice, these agents are often administered as combinations of three to six compounds, commonly referred to as “cocktail therapy.” Although this approach may provide modest symptomatic relief, its overall effectiveness remains limited, and the composition of such regimens varies considerably without standardized protocols.
Condition-targeted interventions include idebenone for Leber hereditary optic neuropathy (LHON) and L-carnitine and arginine/citrulline supplementation for MELAS to reduce stroke-like episodes [65,66]. Symptomatic therapies, including antiepileptic drugs, cardiac pacing, and physiotherapy, are essential for managing neurological, cardiac, and muscular manifestations [61,65]. While the clinical use of these strategies is guided by current insights into mitochondrial disease pathophysiology, the supporting evidence for their therapeutic efficacy remains limited. Table 2 summarizes the conventional and current therapeutic strategies for mitochondrial diseases.
Despite these supportive strategies, the overall prognosis of mitochondrial diseases remains poor, particularly in pediatric cases [76,77]. A systematic review of the natural history of these disorders reported that most cases begin in childhood, with 59% presenting before 18 months of age and 81% before 18 years. Mortality is high, with 13% of patients dying before 1 year, 57% before 5 years, and 74% before 10 years [76]. A retrospective observational study of neonatal mitochondrial diseases found neonatal onset in 28.7% of cases, with an overall mortality rate of 44.8%. Mortality was especially high in patients with cardiomyopathy (60.5%) compared to those with Leigh syndrome (23.1%). The median survival time was only 1.86 years, with a one-year survival rate of 51.8% [77]. These findings underscore the urgent need for effective therapies that move beyond symptomatic management to directly target the underlying defects to restore mitochondrial function. Among the emerging strategies, mesenchymal stromal cell (MSC)-based and MSC-derived extracellular vesicle (MSC-EV)-based therapies have gained significant attention for their potential to achieve such restorative effects, as discussed in the following sections.
5. Therapeutic Potential of Mesenchymal Stromal Cells and Their Extracellular Vesicles in Mitochondrial Dysfunctions
In recent decades, stromal cell therapy has emerged as a promising therapeutic approach. Mesenchymal stromal cells (MSCs) derived from various sources possess several advantageous properties, including low immunogenicity, potent paracrine signaling, antioxidant effects, and the unique ability to migrate to sites of injury and transfer mitochondria [9]. A growing body of evidence from in vitro, in vivo, and clinical studies has consistently demonstrated the therapeutic potential of MSCs across a wide range of degenerative and metabolic disorders, such as diabetes mellitus [78], osteoarthritis [79], wound healing [11], degenerative disc disease [80], and retinal degenerative disease [81]. A recent clinical trial on type-1 diabetes reported that MSC transplantation was safe, improved HbA1c and C-peptide levels, and shifted pro-inflammatory cytokines toward an anti-inflammatory profile [78]. Similarly, a systematic review evaluating MSC therapy for osteoarthritis (OA) concluded that MSCs are safe and effective in reducing pain and improving knee function in patients with knee OA [82]. Furthermore, MSC-derived exosomes—specialized extracellular vesicles—have shown superior therapeutic efficacy compared with MSCs themselves in models of cardiac ischemia–reperfusion injury and diabetic wound regeneration [11,12].
Taken together, these findings highlight the broad therapeutic potential of MSCs and their extracellular vesicles (MSC-EVs) across diverse disease models. Although current evidence underscores their regenerative and protective roles, the application of MSC-based therapies in the context of mitochondrial diseases remains relatively limited. Therefore, the following sections of this review summarize the available in vitro and in vivo studies investigating the therapeutic effects of MSCs and MSC-EVs in mitochondrial diseases.
5.1. Therapeutic Potential of Mesenchymal Stromal Cells in Mitochondrial Dysfunctions
Mesenchymal stromal cells have gained increasing attention for their ability to repair cellular damage and restore mitochondrial function through mechanisms such as paracrine signaling, antioxidant activity, and mitochondrial transfer. Although research on MSC-based therapies for mitochondrial diseases remains limited and is still emerging, it demonstrates promising therapeutic potential. Liu et al. investigated the effects of bone marrow-derived mesenchymal stromal cells (BM-MSCs) and highly purified mesenchymal stromal cells (RECs) in iPSC-derived neurons from patients with MELAS. Both direct and indirect co-culture with MELAS neurons significantly restored mitochondrial function, primarily through mitochondrial transfer from MSCs [83]. This intervention improved mitochondrial membrane potential, ATP production, ROS regulation, intracellular calcium homeostasis, and oxygen consumption rate. The transfer of intact mitochondria and/or mitochondrial components was mediated by tunneling nanotubes (TNTs), connexin-43-mediated gap junction channels (Cx43-GJCs), and extracellular vehicles (EVs) [83].
In another study, co-culture of BM-MSCs with complex I-deficient fibroblasts revealed a significant improvement of mitochondrial respiration, reduced cellular ROS levels, and upregulation of antioxidant enzymes such as SOD2 (superoxide dismutase 2, mitochondrial) and HO-1 (heme oxygenase 1) [84]. Notably, repeated co-culture with MSCs at two sequential time points further enhanced the therapeutic effects, with a more sustained reduction in ROS levels observed after direct co-culture compared to treatment using MSC-conditioned medium alone. Furthermore, co-culture of iPSC-derived neural progenitor cells (NPCs) from an LHON patient with MSCs led to marked mitochondrial genomic improvements, including an increased ratio of wild-type to mutant mitochondrial DNA (mtDNA), elevated levels of the wild-type mtND4 (m.11778A allele), and reduced expression of the mutant mtND4 (m.11778A>G allele) [85]. These genomic shifts were accompanied by a significant enhancement in mitochondrial metabolic function in LHON-derived neurons.
Mitochondrial dysfunction is a central contributor to the pathogenesis of diabetic nephropathy [51], and repeated administration of MSCs has been shown to ameliorate tubular epithelial cell injury and delay disease progression by enhancing mitochondrial function and modulating inflammatory responses [86,87]. Extending these findings to neurological disorders, cerebral ischemia/reperfusion (I/R) injury—a condition marked by severe neuroinflammation and mitochondrial dysfunction [88]—was shown to benefit from treatment with ischemic-hypoxic preconditioned olfactory mucosa-derived MSCs. This treatment exerted significant neuroprotective effects in vitro and in vivo, including reduced infarction, improved motor outcomes, and preservation of mitochondrial integrity [89]. Similar therapeutic benefits have been reported in retinal degenerative disease, where mitochondrial dysfunction and Müller cell gliosis are major pathological features [90]. Treatment of Müller cells with BM-MSCs enhanced mitochondrial function, reduced oxidative stress and gliosis, and partially preserved visual function in degenerative rat retinas [91]. These effects may be attributed to increased mitochondrial DNA (mtDNA) content and the promotion of mitochondrial fusion in damaged Müller cells. Further details of these studies are presented in Table 3.
5.2. Therapeutic Potential of MSC-Derived Extracellular Vesicles in Mitochondrial Dysfunctions
Although many studies have highlighted the regenerative potential of MSC-derived extracellular vesicles, recent investigations have increasingly emphasized their role in alleviating mitochondrial dysfunction, particularly in secondary mitochondrial disorders. Intervertebral disc degeneration (IVDD), whether age-related or induced by genetic and mechanical factors, is strongly associated with mitochondrial impairment, which promotes excessive ROS production, leading to extracellular matrix degradation, loss of nucleus pulposus cells (NPCs), and enhanced inflammatory signaling [92,93,94,95]. In vitro studies demonstrated that umbilical cord MSC-exosomes (UC-MSC-exos) enhance NPC viability, reduce intracellular and mitochondrial ROS, and restore mitochondrial membrane potential, while in vivo evidence shows their ability to slow IVDD progression in rat models [94,95]. Similarly, mitochondrial dysfunction plays a central role in the pathogenesis of kidney disease [96], in which MSC-EVs have been shown to attenuate mitochondrial injury by inhibiting fission, enhancing antioxidant defenses, and promoting ATP production [97]. Notably, hypoxia-preconditioned EVs (Hypo-EVs) exhibit superior therapeutic efficacy in restoring renal function and reducing fibrosis [98]. Further studies demonstrating the mitochondrial-enhancing effects of MSC-EVs are summarized in Table 3.
Therapeutic efficacy evidence of MSC-EVs in genetically defined PMDs is currently very limited. A recent landmark study demonstrated that mitochondria-enriched MSC-EVs (EV-Mito) restored mitochondrial membrane potential, ATP production, and oxidative phosphorylation while reducing mitochondrial reactive oxygen species in Leber hereditary optic neuropathy (LHON) mtDNA-mutant cells and animal models [99]. Collectively, these findings highlight a critical research gap in the field, underscoring the need for further investigation of MSC-EV-based therapies in additional genetically defined primary mitochondrial diseases. While promising therapeutic efficacy has been demonstrated in LHON models, it remains unclear whether MSC-EV-mediated mitochondrial transfer and bioenergetic rescue can be generalized to other PMDs, such as Leigh syndrome, MELAS, or mitochondrial DNA depletion syndromes. Therefore, systematic preclinical studies in diverse PMD models are required to validate the therapeutic potential, disease specificity, and mechanistic efficacy of MSC-EVs as a universal mitochondrial-targeted regenerative strategy.
HF-MSC-Exos; hair follicle mesenchymal stem cell-derived exosomes, Hy-Exos; hypoxic exosomes, Nor-Exos; normoxic exosomes, Nor-EVs; normoxic extracellular vesicles, Hy-EVs; hypoxic extracellular vesicles, BM-MSC; bone marrow mesenchymal stem cells, MELAS; mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episode, AdMSC; adipocyte-derived mesenchymal stem cells, ALI; acute lung injury, UC-MSC; umbilical cord mesenchymal stem cells, IVDD; Intervertebral disc degeneration, COL2A1; collagen type II alpha-1, MMP; mitochondrial membrane potential, TECs; tubular epithelial cells, OM-MSCs; olfactory mucosa mesenchymal stem cells, IhOM-MSCs; Ischemic-hypoxic primed OM-MSCs, POI; premature ovarian insufficiency, AKI; acute kidney injury, SOD2; superoxide dismutase 2, HO-1; heme oxygenase 1, P-MSC-EV; placenta-derived MSC-EV, CKD; chronic kidney diseases.
5.3. Summary of Experimental Evidence on MSC- and MSC-EV-Mediated Restoration of Mitochondrial Function
Mesenchymal stromal cells from various tissue sources—including bone marrow, umbilical cord, adipose tissue, and olfactory mucosa—have shown strong potential in restoring mitochondrial function across multiple disease models [85,86,87,89,91,95,102,103,104,105]. The primary mechanism underlying these effects involves the transfer of functional mitochondria from MSCs to damaged cells, leading to an increased proportion of healthy versus dysfunctional mitochondria [43,85,105]. These healthy mitochondria shift cellular metabolism from glycolysis toward oxidative phosphorylation, thereby increasing mitochondrial membrane potential, enhancing ATP production, and reducing oxidative stress [43]. MSCs also exert potent antioxidant effects by upregulating enzymes such as SOD2 and HO-1, and they promote anti-apoptotic activity through increased expression of Bcl-2 and decreased expression of pro-apoptotic proteins, including BAX, IL-1β and IL-18 [89]. In addition, MSCs exhibit strong anti-inflammatory effects by suppressing M1 macrophage polarization and downregulating inflammatory mediators such as the NLRP3 inflammasome, ASC, caspase-1, caspase-8 and GSDMD [86,89]. Furthermore, MSCs provide mitochondrial protection by upregulating GRP78, a key regulator of mitochondrial homeostasis that maintains the balance between fusion and fission, prevents excessive Ca^2+^ accumulation, reduces free radical production, preserves mitochondrial membrane potential, and sustains respiratory activity [106,107]. Through these combined mechanisms, MSCs mitigate oxidative injury, suppress inflammation, restore cellular bioenergetics, and promote tissue regeneration in both metabolic and degenerative disorders associated with mitochondrial dysfunction.
Similar to their parent mesenchymal stromal cells, MSC-EVs enhance mitochondrial function by restoring mitochondrial membrane potential and increasing ATP synthesis, while concurrently reducing oxidative damage and apoptosis. MSC-EVs also promote tissue protection through modulation of redox homeostasis, cellular metabolism, and pro-survival signaling pathways. Across various disease models, MSC-EVs have been shown to improve organ function, attenuate tissue fibrosis, and facilitate recovery of normal physiological activity by stabilizing mitochondrial dynamics and preventing progressive damage [94,95,97,98,99,100,101,102,103,104]. Furthermore, mitochondria-enriched extracellular vesicles (ME-EVs) have been developed as a cell-free alternative to MSC transplantation and were shown to exhibit no detectable systemic or retinal toxicity in vivo, whereas MSC transplantation was associated with retinal structural alterations, inflammatory responses, and reduced durability of therapeutic effects, supporting the improved safety profile and stability of EV-based therapy [99]. Collectively, these findings support MSC-EVs as a promising cell-free therapeutic platform that preserves much of the mitochondrial and regenerative efficacy of their parent cells while offering improved safety and stability profiles.
Mechanistically, MSC-EVs mediate their effects through the transfer of bioactive molecules—including proteins and nucleic acids—that stimulate mitochondrial homeostasis and antioxidant defense. In an in vitro model of cardiac hypertrophy, MSC-EVs delivered DJ-1, a cytoprotective protein that defends cardiomyocytes against oxidative stress, maintains mitochondrial integrity and inhibits cell death during ischemia–reperfusion injury (IRI) [108]. Transferred DJ-1 enhanced the expression of antioxidant-related proteins such as NRF2, HO-1, SOD2 and GPX4, while reducing NOX4 expression [100]. Moreover, MSC-EVs increased the expression of SIRT3, TFAM, AMPKα, and PGC-1α, which together form a regulatory network that suppresses oxidative stress and enhances cellular energy production [101]. Similar to their parent cells, MSC-EVs exert anti-apoptotic effects by downregulating caspase-3, caspase-8, caspase-9, BAX, and P53, and exhibit anti-inflammatory actions through the suppression of NF-κB, TNF-α, and IL-1β, along with upregulation of IL-4 and IL-10 [101,102,103]. MSC-EVs also promote mitochondrial fusion by increasing the expression of MFN1, MFN2, and OPA1. Furthermore, under stress conditions, MSC-EVs reduce the expression of HSP60, TOMM20, Drp1 and Fis1, while increasing rhot1 expression to regulate cellular responses to stress and maintain mitochondrial homeostasis [102]. Regulatory miRNAs—such as miR-214-3p, from HF-MSC-exosomes—have been shown to inhibit the PI3K/AKT/mTOR pathway, thereby alleviating mitochondrial dysfunction and oxidative stress. Additional signaling pathways, including the p38 and ERK1/2 pathways, are also modulated by MSC-EV bioactive components.
Emerging evidence indicates that priming and engineering strategies can substantially enhance the functional quality and therapeutic efficacy of MSC-derived extracellular vesicles. Beyond indirect paracrine effects, MSC-EVs have also been shown to mediate direct mitochondrial transfer [99,108]. In a recent study, engineered large mitochondria-enriched extracellular vesicles (Super-EV-Mito) efficiently delivered functional mitochondria and restored mitochondrial bioenergetics in cellular and animal models of mitochondrial disease, highlighting EV size and engineering as critical determinants of effective mitochondrial delivery [99]. Mitochondria-enriched EVs, particularly from iPSC-derived cardiomyocytes, have shown a reliable ability to enhance ATP generation, reduce oxidative stress, and reverse functional impairment in damaged cardiac tissue [109]. Moreover, accumulating evidence indicates that hypoxia-primed MSC-EVs exhibit enhanced mitochondrial rescue compared with EVs derived from normoxic MSCs, suggesting that hypoxic priming enriches bioactive cargo and augments the therapeutic potency of MSC-EVs [98,100,101,103]. Consistently, Vitale et al. demonstrated that hypoxia-conditioned MSCs release functionally active EVs capable of promoting tissue remodeling and supporting repair across multiple injured organs, including the brain, liver, spinal cord, kidney, and heart [110,111]. Collectively, MSC-EVs restore mitochondrial health by integrating antioxidant, anti-apoptotic, anti-inflammatory, and metabolic reprogramming mechanisms, offering an effective therapeutic strategy for conditions driven by mitochondrial dysfunction.
Figure 2 illustrates the mechanisms through which MSCs and MSC-EVs may exert their mitochondrial therapeutic effects.
6. Challenges and Prospects of MSC- and MSC-EV-Based Therapies for Mitochondrial Dysfunctions
Although growing evidence indicates that MSCs and MSC-EVs can restore mitochondrial function and hold therapeutic promise for mitochondrial disorders, several challenges must be addressed before these approaches can be translated into practical clinical treatments. Current studies demonstrate that both MSCs and their extracellular vesicles contribute significantly to the restoration of mitochondrial function in secondary mitochondrial dysfunction (SMD), primarily through direct mitochondrial transfer or the delivery of mitochondrial components. Because SMDs commonly arise as a consequence of chronic diseases, inflammatory conditions, or aging, the antioxidative, anti-apoptotic, and anti-inflammatory properties of MSCs and MSC-EVs position them as promising candidates for reversing disease progression and improving mitochondrial function and tissue repair [86,87,89,95]. However, data specific to primary mitochondrial diseases remains limited. Existing investigations have largely focused on MSC-based interventions, with very limited studies to date evaluating the therapeutic potential of MSC-EVs in this context, partly because conventional small extracellular vesicles, including exosomes, have limited capacity to encapsulate and deliver intact mitochondria. This gap raises important questions: Can MSC-EVs reproduce the mitochondrial restorative effects observed with MSCs in primary mitochondrial disorders? And do their bioactive cargos have the capacity to enhance antioxidant defenses, ATP production, and mitochondrial biogenesis in genetically impaired cells? Can EV engineering enhance their capacity to carry intact mitochondria?
Furthermore, available data show that MSCs provide a clear functional rescue in mitochondrially impaired cells by delivering healthy mitochondria and wild-type mtDNA, resulting in reduced oxidative stress and mitigation of downstream dysfunction. Although these findings offer strong evidence of metabolic support and restoration of cellular homeostasis, no correction of the underlying mtDNA or nDNA mutations has been observed. The absence of genomic repair indicates that the therapeutic effect is compensatory rather than curative, which may limit the long-term durability of responses in disorders driven by persistent genetic defects. Future therapeutic strategies may integrate MSC- or MSC-EV-based mitochondrial support with targeted gene therapy to correct underlying mtDNA or nDNA mutations. In this integrated approach, MSCs or their EVs would provide rapid metabolic rescue through delivery of healthy mitochondria and bioactive cargos, whereas gene-editing platforms (e.g., CRISPR-based systems or mito-targeted nucleases) would ensure durable correction of the primary defect. Such a combinatorial strategy may overcome the limitations of purely compensatory therapies and advance MSC/MSC-EV applications from transient support toward truly disease-modifying interventions in mitochondrial disorders.
A growing body of evidence indicates that mesenchymal stromal cells can restore mitochondrial function by transferring healthy mitochondria to damaged cells [85,105]. However, the precise mechanisms underlying these therapeutic effects remain incompletely understood and require further exploration. Critical gaps include whether the transferred mitochondria act independently to supply ATP and counter oxidative stress, whether they fuse with endogenous dysfunctional mitochondria to restore network integrity, or whether they stimulate mitophagy to clear damaged organelles. It also remains unknown whether these donated mitochondria can replicate and sustain long-term function, a process that requires coordinated mtDNA replication and nuclear-encoded protein import. The durability of their effects within defective cells likewise remains an open question. In parallel, MSC-derived EVs have shown the ability to improve mitochondrial function, yet the mechanisms responsible for these effects are still in need of exploration and discussion. Key questions include whether MSC-derived extracellular vesicles can effectively transfer intact mitochondria under physiological conditions or whether this process requires induction through priming or bioengineering strategies, and how priming conditions or MSC source influence mitochondrial cargo composition and functionality. Together, these unresolved issues highlight several priority areas for immediate future investigation, including the fate and functional integration of transferred mitochondria, the molecular composition of MSC-EV mitochondrial cargo, and the temporal stability of these therapeutic effects in impaired cells.
Beyond mechanistic uncertainties, manufacturing constraints—particularly those related to cell production and scalability—also limit clinical translation. MSC expansion is limited by finite passage numbers, donor variability, senescence, and increasing batch heterogeneity, all of which restrict large-scale manufacturing and complicate regulatory compliance. To address these challenges in MSC production, recent studies have introduced a pluripotent stem cell-based method for MSC induction. Human pluripotent stem cell-derived MSCs exhibit superior expansion capacity (exceeding 30 passages), enhanced proliferative potential, resistance to senescence, and robust secretory profiles, including increased cytokine and exosome production [112]. Moreover, the inherent heterogeneity of MSC populations complicates standard culture practices, making them inefficient and resource-intensive. To overcome this limitation, recent advances have led to the development of high-purity MSCs known as rapidly expanding clones (RECs), derived from single MSCs. These RECs demonstrate improved homogeneity, superior mitochondrial integrity, and a stronger capacity to restore bioenergetics and mitochondrial function in deficient cells [113]. In contrast, EVs can be produced from standardized MSC sources with high scale, greater batch-to-batch consistency and reduced biological variability [65,69]. Moreover, EVs demonstrate superior stability compared with MSCs; they tolerate multiple freeze–thaw cycles, maintain bioactivity during long-term storage, and can be formulated as off-the-shelf medicinal products without the viability constraints or complex cryopreservation requirements associated with living cells [112,113]. These attributes significantly reduce production costs, simplify storage and transport logistics, and facilitate the development of standardized, scalable EV-based therapies.
Advances in production must be accompanied by rigorous evaluation of safety. Although several studies suggest that MSC- and MSC-EV-based therapies are generally well tolerated, their long-term safety profile remains unclear. Existing clinical and preclinical reports indicate minimal immune reactivity and a low risk of tumorigenicity [114]. For example, Vega-Letter et al. demonstrated that xenogeneic mitochondrial transplantation did not elicit adaptive immune responses and even exerted anti-inflammatory and tissue-protective effects in osteoarthritis models [115]. Similarly, the Stem Cell Ophthalmology Treatment Study (SCOTS), which evaluated MSC therapy for mitochondrial optic neuropathies, reported no adverse events during a 12-month follow-up period [116,117,118,119]. However, such short observation windows cannot exclude delayed adverse effects, genomic instability, or long-term immunological consequences. In this context, MSC-derived extracellular vesicles may offer a comparatively safer alternative to whole-cell therapy. MSC-EVs exhibit markedly lower immunogenicity and negligible tumorigenic potential while preserving many of the therapeutic functions of their parent cells. They can suppress T- and B-cell proliferation, modulate macrophage activity, inhibit NK-cell responses, and reduce immune rejection [120,121]. Nevertheless, despite these advantages, comprehensive long-term safety assessments remain lacking for both MSCs and MSC-EVs, underscoring the need for extended follow-up studies and standardized safety monitoring in future clinical applications.
Another obstacle for clinical translation of MSC- and MSC-EV-based therapies is the complexity of primary mitochondrial disorders, which often involve multisystem dysfunction. Although MSCs have demonstrated the ability to home to distant organs such as the brain, heart, lungs, and kidneys [122], it remains uncertain whether this capacity is sufficient for diseases that affect multiple organs simultaneously. This may raise pivotal questions: Will the homing efficiency of MSCs and MSC-EVs be adequate in multi-organ contexts? Can the same dosing strategies, preparation methods, and routes of administration used in single-organ involvement in SMDs be applied to PMDs? Addressing these questions will require robust in vivo disease models to determine optimal dosage, delivery routes, and treatment duration, as well as to evaluate therapeutic efficacy and biodistribution.
Taken together, the therapeutic application of MSCs and MSC-EVs in mitochondrial dysfunctions remains an active and critically important area of investigation. Emerging technologies in cell-free regenerative medicine—such as bioengineered EVs and hypoxia priming—are expected to enhance therapeutic efficiency, safety, and scalability. Future directions should focus on:
- Defining the molecular mechanisms of mitochondrial repair mediated by MSCs and EVs.
- Developing standardized protocols for MSC/EV production, storage, and administration.
- Establishing multi-organ disease models to evaluate homing capacity, dosing requirements, and biodistribution.
- Conducting long-term safety and efficacy studies to assess immune tolerance and functional recovery.
With continued refinement and validation, MSCs and their EVs may provide a viable platform for developing targeted therapies for mitochondrial diseases, helping to narrow the gap between current symptomatic management and future mechanism-based treatments.
7. Conclusions
Mitochondrial dysfunction represents a major barrier to cellular homeostasis and contributes to a wide range of degenerative, inflammatory, and genetic disorders. Growing evidence demonstrates that mesenchymal stromal cells and their extracellular vesicles can restore mitochondrial function through mechanisms that include mitochondrial transfer, delivery of mitochondrial components, and modulation of oxidative stress and inflammatory signaling. Although these findings highlight the therapeutic promise of MSC- and MSC-EV-based interventions, the precise mechanisms governing their actions, durability, and integration within defective cells remain incompletely understood. Furthermore, despite encouraging short-term safety data, long-term safety and immunological consequences of both approaches require systematic investigation. MSC-EVs—with their lower immunogenicity, greater stability, and scalable production—represent a compelling cell-free alternative to MSC therapy; however, their mitochondrial cargos and mechanistic pathways remain to be fully elucidated. Overall, MSCs and MSC-EVs hold significant potential as next-generation therapeutics for mitochondrial disorders, but translating this promise into clinically effective, durable, and safe treatments will require closing existing knowledge gaps and advancing both mechanistic understanding and regulatory-ready production platforms.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Al Ojaimi M. Salah A. El-Hattab A.W. Mitochondrial fission and fusion: Molecular mechanisms, biological functions, and related disorders Membranes 20221289310.3390/membranes 1209089336135912 PMC 9502208 · doi ↗ · pubmed ↗
- 2Spinelli J.B. Haigis M.C. The multifaceted contributions of mitochondria to cellular metabolism Nat. Cell Biol.20182074575410.1038/s 41556-018-0124-129950572 PMC 6541229 · doi ↗ · pubmed ↗
- 3El-Hattab A.W. Suleiman J. Almannai M. Scaglia F. Mitochondrial dynamics: Biological roles, molecular machinery, and related diseases Mol. Genet. Metab.201812531532110.1016/j.ymgme.2018.10.00330361041 · doi ↗ · pubmed ↗
- 4Niyazov D.M. Kahler S.G. Frye R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment Mol. Syndromol.2016712213710.1159/00044658627587988 PMC 4988248 · doi ↗ · pubmed ↗
- 5Gorman G. Chinnery P. Di Mauro S. Hirano M. Koga Y. Mc Farland R. Suomalainen A. Thorburn D.R. Zeviani M. Turnbull D.M. Mitochondrial Diseases Nat. Rev. Dis. Primers 201621608010.1038/nrdp.2016.8027775730 · doi ↗ · pubmed ↗
- 6Rambani V. Hromnikova D. Gasperikova D. Skopkova M. Mitochondria and Mitochondrial Disorders: An Overview Update Endocr. Regul.20225623224810.2478/enr-2022-002535843711 · doi ↗ · pubmed ↗
- 7Zhao X. Yu M. Zhang W. Hou Y. Yuan Y. Wang Z. Demographic Characteristics, Diagnostic Challenges, Treatment Patterns, and Caregiver Burden of Mitochondrial Diseases: A Retrospective Cross-Sectional Study Orphanet J. Rare Dis.20241928710.1186/s 13023-024-03289-539095827 PMC 11297657 · doi ↗ · pubmed ↗
- 8Ahuja A.S. Understanding Mitochondrial Myopathies: A Review Peer J 20186 e 479010.7717/peerj.479029844960 PMC 5967365 · doi ↗ · pubmed ↗