Genetic Mapping Identifies Stable QTL and Candidate Genes Regulating Internode Proportion for Maize Plant Architecture Improvement
Xueying Li, Hao Zhang, Keying Wan, Xiaoqian Qiu, Qiankun Xie, Geming Guo, Yuehua Zhao, Zibo Ding, Xiaoyang Chen, Hongyu Chen, Huiling Xie, Jihua Tang, Xuehai Zhang, Dong Ding

TL;DR
This study identifies genetic regions and genes that influence the structure of maize plants, which could help breeders improve crop yield.
Contribution
The study discovers stable QTL and candidate genes related to maize internode proportion using genome-wide association analysis.
Findings
821 significant SNPs and 417 QTL were identified for traits related to maize internode proportion.
Four stable QTL (qU1–qU4) were identified through colocalization analysis.
Two candidate genes were supported by haplotype analysis and showed significant differences in internode-related traits.
Abstract
Background: Ideal plant architecture is central to high-yield maize breeding. The proportional length of internodes above the ear plays a crucial role in determining plant architecture. Methods: In this study, we used an association panel comprising 288 maize inbred lines and performed genome-wide association studies (GWASs) with 1.25 M high-density single nucleotide polymorphism (SNP) markers under a Q + K mixed linear model. Results: A total of 821 significant SNPs associated with plant height (PH), height above ear (HAE), and internode-related traits were detected, which were further classified into 417 quantitative trait loci (QTL). Among these, 128 significant SNPs and 44 QTL were identified for the U1/HAE trait, and 37 significant SNPs and 27 QTL for the U1/PH trait. Four stable QTL (qU1–qU4) were identified through colocalization analysis. Two candidate genes, Zm00001d013222…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
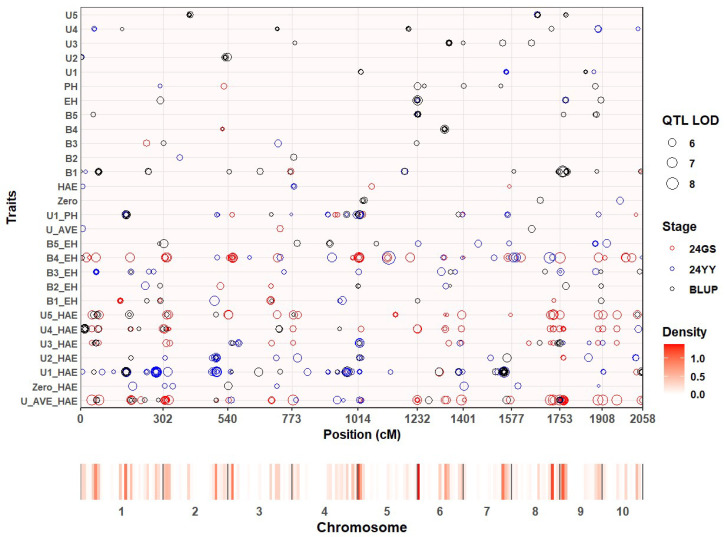 Figure 1
Figure 1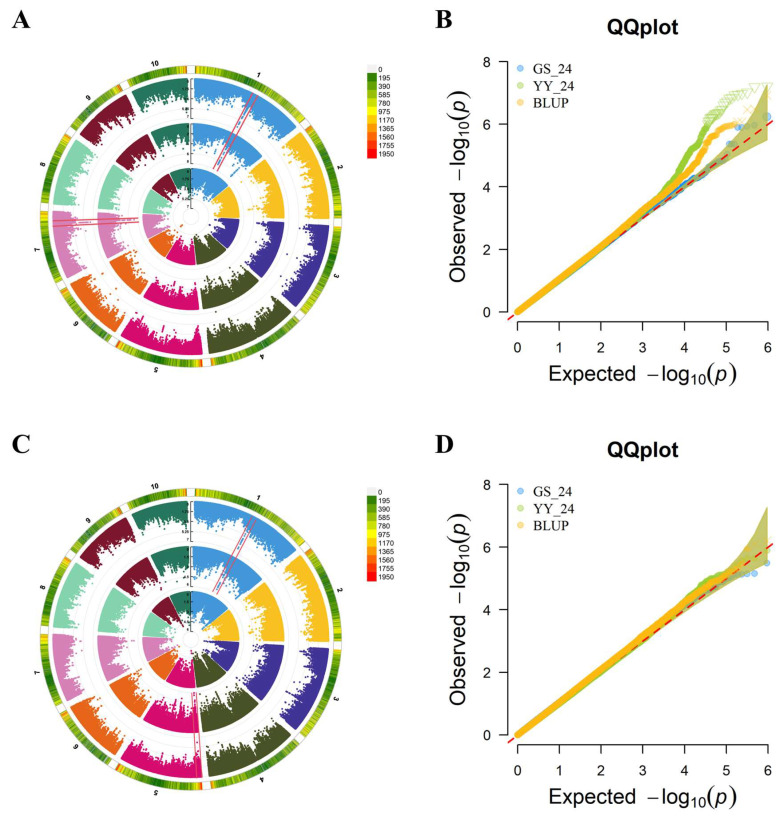 Figure 2
Figure 2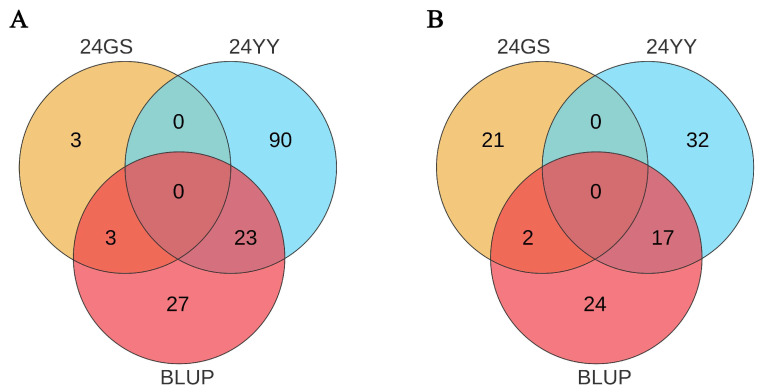 Figure 3
Figure 3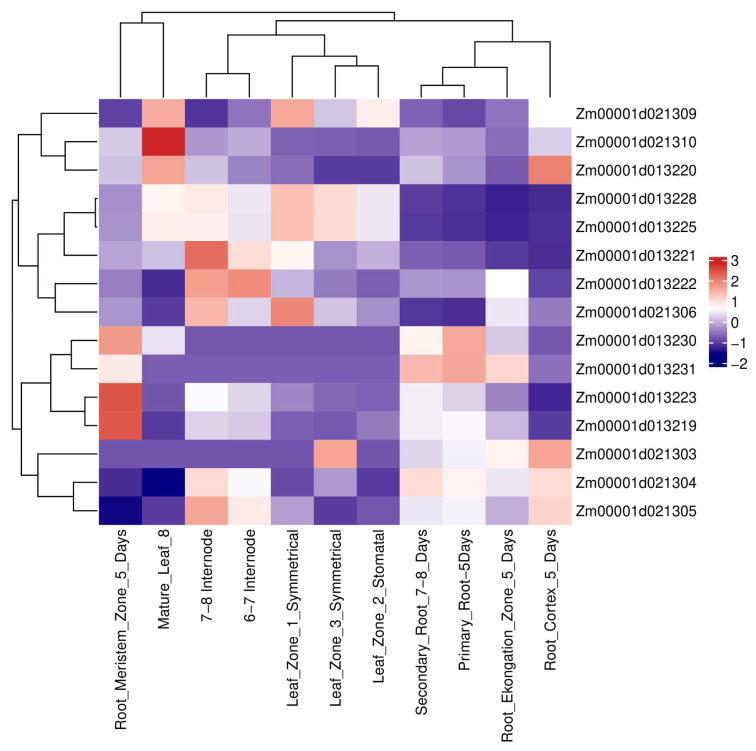 Figure 4
Figure 4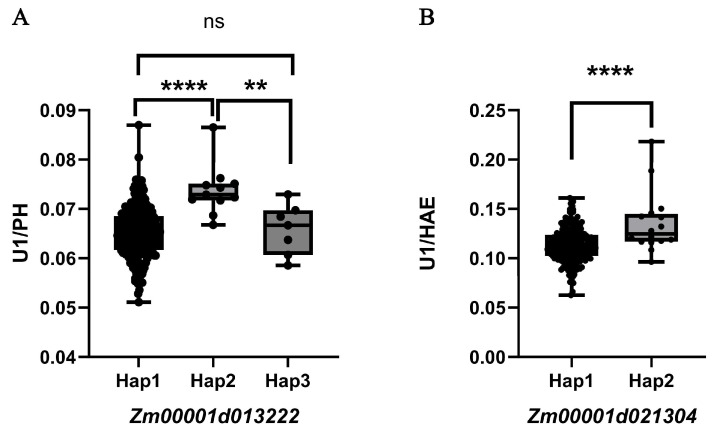 Figure 5
Figure 5- —National Natural Science Foundation of China
- —Natural Science Outstanding Youth Fund of Henan Province
- —Key research and development projects of Henan Province
- —Joint Foundation for Science and Technology Research and Development Program of Henan Province
- —Henan Province Science and Technology Attack Project, China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Mapping and Diversity in Plants and Animals · Crop Yield and Soil Fertility · Greenhouse Technology and Climate Control
1. Introduction
Plant height (PH) and internode length (IL) are key determinants of ideal plant architecture and are closely associated with crop yield and plant adaptability. In maize, optimizing plant architecture has become a major focus in breeding programs to enhance productivity and resilience under varying environmental conditions [1]. As one of the world’s most important cereal crops, maize is frequently subjected to abiotic stresses that can compromise structural stability and ultimately limit yield [2].
Genetic studies have identified several genes that regulate maize internode elongation and plant stature. For example, the dwarf gene Brachytic2 (br2) shortens lower internodes and alters stem morphology [3], while its allele qpa1 reduces PH and ear height (EH) and increases stem thickness [4]. The gif1 gene promotes cell proliferation in stems, and its mutant exhibits shortened internodes [5]. Similarly, transcription factors such as BLH12 and BLH14 interact with KNOTTED1 to maintain meristem activity and ensure normal stem elongation [6]. These findings underscore the genetic complexity underlying internode development and its importance in shaping plant architecture.
Internode elongation is coordinately regulated by a network of hormone signaling pathways, including auxin, gibberellin (GA), brassinosteroid (BR), ethylene, jasmonic acid (JA), and strigolactone (SL) [7]. For instance, the GRAS42 gene modulates internode development through BR-mediated signaling and cell wall biosynthesis [8]. Mutants of GRAS42 exhibit a dwarf phenotype, with plant height reduced to approximately 60% of the wild type due to shortened internodes [9]. This effect is further enhanced in the nana plant1-1 and gras42-mu1021149 double mutant, underscoring the gene’s role in internode regulation [9]. Similarly, disrupted GA signaling inhibits internode elongation and reduces plant height [10], while the jasmonate analog Coronavirin suppresses maize internode elongation by downregulating the cell wall-associated gene ZmXTH1 [11]. Ethylene also contributes to this process, as evidenced by the ZmACS7 gene, which influences the elongation of both internode and auricle cells [12]. Additionally, strigolactone biosynthesis is critical for normal stem development; mutations in ZmCCD8, which encodes a carotenoid cleavage dioxygenase, impair SL production and lead to reduced stem diameter, compromised internode elongation, and overall plant dwarfism [13]. Collectively, these findings illustrate the sophisticated hormonal crosstalk that integrates multiple pathways to coordinately control stem growth and architecture.
Despite advances in understanding PH genetics, the genetic basis of specific internode proportions—particularly those above the ear—remains underexplored. Traditional studies often focus on overall PH or ear height, yet the proportional length of individual internodes may offer more precise targets for architectural improvement. For example, the first internode above the ear (U1) has been noted for its structural importance and susceptibility to environmental stress [14,15]. Studies have demonstrated that knockout of the ZmD1 gene suppresses longitudinal elongation while promoting transverse expansion of internode cells, leading to reduced plant height and shortened internodes [16]. The ZmTE1 gene plays a crucial role in maintaining internode meristem formation and cell division and regulates internode elongation by promoting cell extension [17]. Notably, in maize breeding research, three quantitative trait loci (QTL) associated with internode length have been identified, from which 303 related genes have been annotated [18]. These genes influence internode elongation primarily by mediating phytohormone signaling, modulating receptor activity, and regulating carbon metabolism pathways. Among them, ZmIL1, ZmIL2, and ZmIL3 have been highlighted as key candidate regulators of internode length [18].
In this study, we employed a GWAS approach using a diverse association panel of 288 inbred lines genotyped with 1.25 M SNPs to dissect the genetic architecture of internode proportion traits, with a particular focus on U1/HAE and U1/PH. Our objectives were to (1) characterize phenotypic variation in internode-related traits across two environments; (2) identify significant SNPs and stable QTL associated with internode proportions; and (3) pinpoint candidate genes through integrated bioinformatic and haplotype analyses. This work provides new insights into the genetic regulation of maize plant architecture and offers candidate loci for molecular breeding.
2. Materials and Methods
2.1. Plant Materials and Experimental Design
The association mapping panel consisted of 288 maize inbred lines, including 131 tropical/subtropical and 157 temperate accessions. In 2024, the panel was planted at two field locations: Yuanyang Modern Agricultural Science and Technology Park (35° N, 113° E; designated as 24YY; average altitude: 74 m; mean annual temperature: 14.3 °C; mean annual precipitation: 550 mm; soil organic matter: 10.6 g·kg^−1^; total nitrogen: 1.1 g·kg^−1^) and Zhangye, Gansu (39° N, 100° E; designated as 24GS; 39° N, 100° E; designated 24GS; average altitude: 1400–1500 m; mean annual temperature: 8.4 °C; mean annual precipitation: 197.4 mm; soil organic matter: 18.0 g·kg^−1^; total nitrogen: 1.1 g·kg^−1^). Each inbred line was replicated twice per location in a full randomized block design, with a row length of 3.0 m, plant spacing of 0.25 m, and row spacing of 0.67 m. Standard field management practices were followed throughout the growing season.
2.2. Phenotypic Data Collection
At physiological maturity, five representative plants per row were selected for phenotyping. Plant height (PH) and ear height (EH) were measured in the field for each plant. Subsequently, whole stalks were harvested and transferred to the laboratory for detailed internode measurements. Internode traits included the length of the ear-position internode (designated Zero), the lengths of the five internodes above the ear (U1–U5), and the five below the ear (B1–B5). (Supplementary Figure S1).
2.3. Phenotypic Data Analysis
For each trait, the mean value per inbred line was first calculated from five plants within each environment. All general statistical analyses in this study were conducted using the spreadsheet program within the WPS Office 12.1.0 software suite (Microsoft Excel-compatible). For maize trait measurements, descriptive statistics were calculated as follows: the mean was computed using the AVERAGE function, skewness and kurtosis were determined with the SKEW and KURT functions, respectively, and the sample standard deviation was derived using the STDEV.S function. The coefficient of variation (CV) was calculated as:
To assess environmental effects, Pearson correlation between traits across the two locations was evaluated using the cor() function in R (v4.3.1). Best linear unbiased predictions (BLUPs) for each trait across the two environments (24GS and 24YY) were then estimated using the lme4 package, with genotype as a random effect and environment as a fixed effect, the broad-sense heritability (h^2^) for each trait was calculated using the following formula:
where is the genotypic variance, is the genotype × environment interaction variance, is the residual error variance, n is the number of environments, and r is the number of replications. Variance components were estimated using analyzed by analysis of variance (ANOVA) using the aov() function in R (v4.3.1). Both the environment-specific line means and the across-environment BLUP were used as phenotypic inputs in the subsequent genome-wide association analysis. Frequency distributions of phenotypic data were visualized with GraphPad Prism 10.1.2.
2.4. Genome-Wide Association Study (GWAS)
Genotype data for the association panel, consisting of approximately 1.25 million (1.25M) single nucleotide polymorphism (SNP) markers aligned to the B73 RefGen_v4 reference genome, along with corresponding population structure information, were obtained from the publicly available accessible dataset provided by Prof. Jianbing Yan of Huazhong Agricultural University. The dataset is available for download at http://www.maizego.org/Resources.html (accessed on 20 January 2026) [19]. After rigorous quality control of each dataset, genotypes from four genotyping platforms (600K, 50K, RNA-seq, and GBS) were merged. Conflicting locus calls were resolved by prioritizing platforms in the order 600K > 50K > RNA-seq > GBS. Genotype imputation was performed using Beagle v4.0 [20]. To optimize imputation parameters and validate accuracy, chromosome 10 markers were used as a reference set [21]. A total of 15,000 known genotypes (approximately 3% of chromosome 10 loci, with three individuals per locus) were randomly masked as missing, and imputation reliability was assessed by comparing the masked true genotypes with the imputed values. Testing multiple parameter combinations identified the optimal settings: window = 50,000, overlap = 5000, ibd = true. Two imputation strategies were compared: the “remove-then-impute” strategy (filtering SNPs with >90% missing rate before imputation) achieved an average accuracy of 96.93%, whereas the “impute-then-remove” strategy (filtering after imputation) yielded a lower accuracy of 95.89% [21]. Accordingly, the former strategy was adopted for final analyses [21]. An integrated genetic map comprising 2.65 million loci was constructed for 540 individuals; of these, 1.25 million loci with minor allele frequency (MAF) ≥ 5% were retained for downstream studies [21]. The final merged HapMap-format genotype dataset is available at www.maizego.org/Resources (accessed on 20 January 2026). GWASs were performed in Tassel 5.2.81 using the Q + K mixed linear model to correct for population structure and kinship. The significance threshold was set at p ≤ 1.0 × 10^−5^ [18]. Result visualization—including Manhattan plots, quantile–quantile (Q-Q) plots, and composite figures—was conducted in RStudio (2025.9.2.0) with the CMplot, ggplot2, ggridges, and corrplot packages.
2.5. Candidate Gene Identification
For each significantly associated SNP, a 100 kb window (50 kb upstream and downstream) was defined as the QTL interval, corresponding to the average linkage disequilibrium decay distance (~50 kb) in this population [22]. Gene annotations within these regions were retrieved from the MaizeGDB database (B73 RefGen_V4, https://maizegdb.org/, accessed on 20 January 2026). Functional descriptions and expression profiles of candidate genes were examined, and expression heatmaps were generated using the Omicshare online platform (https://www.omicshare.com/tools/Home/Task, accessed on 20 January 2026).
2.6. Haplotype Analysis
Haplotype analysis was carried out with Tassel 5.2.81. For candidate genes, gene-based association analysis was performed by extracting gene sequences from the association panel and subsequently evaluating their statistical associations with the target phenotypic traits. Significant SNPs were used to construct haplotypes. Haplotypes represented by fewer than 10 accessions were excluded from further analysis. Phenotypic differences among haplotypes were assessed for statistical significance using a two-tailed t-test.
3. Result
3.1. Phenotypic Variation in Internode-Related Traits
A total of 28 internode-related traits were investigated in this study, including plant height (PH), ear height (EH), height above ear (HAE), lengths of internodes above the ear (U1–U5), lengths of internodes below the ear (B1–B5), the ear-position internode (Zero), the average length of the five internodes above the ear (U_AVE), and a series of proportional traits: U1-U5 to HAE, B1-B5 to EH, Zero to HAE, U_AVE to HAE, and U1 to PH. All traits exhibited considerable phenotypic variation across the association panel. Coefficient of variation (CV) values ranged from 0.02 to 0.28 under the two environments and their best linear unbiased predictions (BLUPs) (Table 1). Skewness and kurtosis values for each trait were generally between −1 and 1, indicating that the phenotypic distributions were approximately normal (Supplementary Figure S2) and consistent with quantitative inheritance controlled by multiple genes. Heritability was estimated for all traits under two environments, and the results showed that the heritability of different traits exhibited significant high–low differentiation characteristics between the two environments. These results confirm that the panel captures broad genetic diversity suitable for genome-wide association mapping of internode architecture.
Notably, within the same environment, internode lengths (ILs) at different positions above the ear were strongly positively correlated (Supplementary Figure S3A–C), suggesting shared genetic regulation or linkage among underlying loci. In contrast, correlations for the same IL trait across different environments were markedly lower (Supplementary Figure S3D–F), highlighting the environmental sensitivity of upper-ear internodes and the dominant role of environment in shaping their phenotypic expression.
To further evaluate the architectural role of internodes above the ear, we examined the proportional contributions of individual internodes. The ratio of the first internode length (U1) to both height above ear (HAE) and plant height (PH) was consistently and significantly higher than that of the other four internodes across environments and BLUPs (Supplementary Figure S4). This stable, dominant proportional role suggests that U1 is a structural cornerstone of the upper-plant profile and likely exerts a major influence on overall above-ear morphology. Given its pronounced and consistent phenotypic contribution [14,15], we focused subsequent genetic analyses on the traits U1/HAE and U1/PH.
3.2. Genome-Wide Association Analysis of Internode-Related Traits
We performed genome-wide association studies (GWASs) on maize internode length (IL) and internode ratio traits using approximately 1.25 million SNP markers and the Q + K mixed linear model. Analyses were conducted separately for each of the two environments (24GS and 24YY) and for across-environment best linear unbiased predictions (BLUPs) (Supplementary Figures S5–S8; Supplementary Table S1). After accounting for population structure and kinship, 821 SNPs exceeded the significance threshold of −log_10_(p) > 5.0 (p < 1 × 10^−5^). These SNPs were grouped into 417 non-redundant quantitative trait loci (QTL) based on a 50 kb linkage-disequilibrium (LD) window. QTL were distributed across all ten maize chromosomes, with notable enrichment on chromosomes 1, 2, 5, and 7 (Figure 1). Among all the identified QTL, 13 stable QTL were screened through colocalization analysis across at least two environments, and these QTL intervals contained a total of 60 internode-related candidate genes (Supplementary Table S2). These associated regions provide a foundation for subsequent candidate-gene identification related to internode proportion.
To further dissect the genetic basis of the first-internode proportion in shaping above-ear architecture, we focused GWASs on the traits U1/HAE and U1/PH. For U1/HAE, 128 significant SNPs were detected across environments and BLUPs (−log_10_(p) > 5.0). Based on the population LD decay distance (~50 kb), these SNPs defined 44 QTL, distributed on chromosomes 1, 2, 3, 4, 5, 6, 7, and 10. Integration of signals from the 24YY environment and the BLUP dataset revealed two consistently detected, stable QTL: qU1 (chr1: 165.32–165.52 Mb) and qU2 (chr7: 148.32–148.51 Mb) (Figure 2A,B). For U1/PH, 37 significant SNPs were identified, corresponding to 27 QTL located on all chromosomes except chromosome 8. Through combined analysis of the 24YY environment and BLUP data, two stable QTL were consistently mapped: qU3 (chr1: 165.38–165.52 Mb) and qU4 (chr5: 6.71–6.81 Mb) (Figure 2C,D). The repeated detection of these QTL across independent datasets indicates that they represent genomic regions with stable genetic effects on internode proportion.
3.3. Analysis of Candidate Genes for U1/HAE and U1/PH Traits
To identify candidate genes underlying U1/HAE and U1/PH, we performed colocalization analysis using phenotypic data from both individual environments and their BLUPs. For U1/HAE, 23 genes were consistently identified between the 24YY environment and BLUPs, and three genes overlapped between 24GS and BLUPs (Figure 3A). For U1/PH, 17 genes overlapped in the 24YY-BLUP comparison, and two genes were shared between 24GS and BLUPs (Figure 3B). Based on peak SNPs from Manhattan plots, we further screened candidate genes within the stable QTL intervals qU1, qU2, qU3, and qU4 (Table 2). No annotated genes were found in the qU1 and qU3 regions. Within the qU2 interval (chr7: 148.32–148.51 Mb), ten candidate genes were identified, including the receptor-like protein kinase Zm00001d021303 (putatively involved in cell signaling), endoglucanase 2 Zm00001d021304 (associated with cell wall polysaccharide degradation), endoplasmic reticulum lumen protein-retention receptor family protein Zm00001d021306, and triosephosphate isomerase Zm00001d021310 (implicated in glycolytic metabolism). The qU4 interval (chr5: 6.71–6.81 Mb) contained thirteen candidate genes, such as gibberellin-regulated protein 10 Zm00001d013222 (a growth-related regulator), the microtubule-associated protein Zm00001d013224 (MOR1), the transcription factor Zm00001d013232 (bHLH113), and arginine N-methyltransferase 2 Zm00001d013219, along with several genes of unknown function. These candidate genes are significantly associated with the proportional traits of the first internode and are implicated in diverse biological processes, including hormone response, cell wall dynamics, signal transduction, and metabolic regulation. Their identification provides a functional roadmap for further elucidation of the genetic networks controlling maize internode proportion and plant architecture.
3.4. Expression Profiling and Haplotype Analysis of Key Candidate Genes
To evaluate the biological relevance of candidate genes underlying U1/HAE and U1/PH, we examined their expression patterns using publicly available transcriptome data (Figure 4). Among the 23 genes located in colocalized intervals, Zm00001d021304 (encoding endoglucanase 2), Zm00001d013221 (a domain-containing protein), Zm00001d013222 (gibberellin-stimulated-like 10), and Zm00001d013223 (retrovirus-related Pol polyprotein) displayed notably higher expression in roots, stems, and leaves relative to other candidates, supporting their potential roles in vegetative growth and development.
Given that internode elongation is coordinately regulated by multiple hormones, we focused on two functionally annotated genes: Zm00001d013222, implicated in gibberellin signaling [23], and Zm00001d021304, involved in cell wall remodeling [24]. Using BLUP phenotypes from both environments, we performed haplotype-based association analysis for these genes (Figure 5; Table 3 and Table 4). For Zm00001d013222, three haplotypes were identified: Hap1 (ATACAC), Hap2 (GCGAGG), and Hap3 (ATACAG). Hap1 and Hap3 did not differ significantly in U1/PH, but both produced significantly lower U1/PH values than Hap2 (p < 0.01). This haplotype-dependent phenotypic variation supports Zm00001d013222 as a candidate regulator of U1/PH. For Zm00001d021304, two haplotypes were found: Hap1 (GGTA) and Hap2 (TTAG). Hap2 exhibited a significantly higher U1/HAE value compared to Hap1 (p < 0.001), confirming Zm00001d021304 as a strong candidate for U1/HAE. These results highlight Zm00001d013222 and Zm00001d021304 as promising genetic determinants of internode proportion, operating through hormone-mediated growth regulation and cell wall dynamics, respectively.
4. Discussion
Currently, with increasing demands for high-density planting in agriculture, maize production faces emerging challenges, such as greater lodging susceptibility, highlighting an urgent need for moderately dwarf hybrid maize varieties [25]. The maize “smart plant architecture” gene lac1, encoding a brassinosteroid C-22 hydroxylase, regulates upper leaf angle [26]. Key QTL controlling compact plant architecture in maize (UPA1/brd1, UPA2/RAVL1) have been successfully cloned, and the introgression of teosinte ligule alleles has been used to modify plant architecture. These improvements reduce resource competition under dense planting and significantly increase maize yield under high-density conditions [27]. In the present study, the ratios of U1 to HAE and to PH remained stable across two environments and BLUP values, and they were significantly higher than those of the other upper internodes. This suggests that modulating the U1 proportion could reduce redundant growth of the upper internodes without altering overall plant height. For example, a moderate reduction in the U1 ratio may shorten the first internode above the ear, lower the plant’s center of gravity, and thereby decrease stalk lodging risk. Previous studies have frequently identified the first internode above the ear as a critical site prone to breakage during stalk failure [14,15]. Moreover, the “U1/HAE” and “U1/PH” ratios represent more stable genetic traits than individual phenotypic measurements. In summary, by focusing on U1 ratio traits, this study not only enriches the genetic understanding of maize plant architecture regulation but also provides directly applicable resources for lodging-resistance breeding through the identification of stable QTL and candidate genes.
Maize internode elongation is coordinately regulated by multiple hormonal pathways. Transcription factors such as ZmABI7 and ZmMYB117 directly bind to the promoters of target genes (ZmCYC1, ZmCYC3, ZmCYC7, and ZmCPP1) to modulate cell cycle progression and cell wall modification, thereby influencing internode development [28]. Gibberellin (GA) shapes maize stem morphology by regulating peroxidase activity and thus cell wall lignification [29]. In rice, ACE1 encodes a protein of unknown function that confers cell division capacity in the intercalary meristem, leading to GA-dependent internode elongation [30]. Another rice gene, PINE1, encodes a zinc-finger transcription factor that reduces stem sensitivity to GA; the floral meristem enhances stem responsiveness to GA by downregulating PINE1 expression [31]. These studies collectively underscore the pivotal role of plant hormones, particularly GA, in internode development. Consistently, among our colocalized candidate genes, Zm00001d013222 encodes a gibberellin-regulated protein 10 (GSL10), which has been implicated in growth and developmental regulation [32]. Based on its functional annotation, we propose that Zm00001d013222 is a key candidate gene regulating the U1/PH trait in this study, likely affecting stem growth and development by modulating gibberellin synthesis or metabolism in maize stems, ultimately leading to alterations in stem morphology.
During stem growth and development, the vascular cambium continuously proliferates, producing new cells. Xylem, which conducts water, differentiates inward from the cambium, while phloem—the main conduit for photoassimilate transport—differentiates outward. The completion of cell division relies on the precise assembly of a new cell wall between daughter cells [33]. Cellulose microfibrils are crucial for the structural organization of plant cell walls, allowing plants to sustain turgor-driven growth habits [34]. Endo-1,4-β-D-glucanases (EGases) are also speculated to function in cell expansion. Although their roles in cell wall synthesis and remodeling are not fully resolved, these enzymes cleave β-1,4-glycosidic bonds in polysaccharides, such as cellulose and xyloglucan [35]. Together, these reports highlight the central importance of the cell wall in stem growth, development, and morphogenesis. In our study, Zm00001d021304 (EG2), a candidate gene colocalized with U1/HAE, encodes an endoglucanase belonging to glycoside hydrolase family 9 (GH9; also termed cellulase). It participates in multiple key processes of cell wall metabolism in higher plants, including the regulation of cellulose biosynthesis and degradation, modification of other (1,4)-β-glucan-containing wall polysaccharides, and mediation of cell wall loosening during cell elongation [24]. Analysis of EG2 expression profiles using the qTeller MaizeGDB platform (https://qteller.maizegdb.org) revealed that its expression peaked 24 days before pollination and remained relatively stable during other periods, suggesting involvement in the stem-jointing stage of maize. EG2 may promote the degradation of cell wall polysaccharides to maintain wall plasticity, thereby facilitating stem elongation. Notably, the previously reported stiff1 gene enhances stalk strength and lodging resistance by negatively regulating cellulose and lignin synthesis and influencing cell wall thickness [36]. This gene may synergize with the identified EG2 gene to balance internode proportion optimization and stem strength enhancement dynamically, a core objective of lodging-resistant plant architecture breeding.
5. Conclusions
Through a GWAS approach in a diverse maize panel, we identified stable QTL (qU1–qU4) and candidate genes governing the proportion of the first internode above the ear. Two functionally annotated genes, Zm00001d013222 (involved in gibberellin signaling) and Zm00001d021304 (a cell wall-modifying endoglucanase), exhibited significant haplotype-based associations with U1/PH and U1/HAE, respectively. These findings highlight the coordinated roles of hormone pathways and cell wall metabolism in shaping maize internode architecture. The stability of these loci across environments underscores their potential value for molecular breeding. This study provides novel genetic insights and precise targets for improving plant architecture and lodging resistance, supporting the development of high-yield maize varieties optimized for modern high-density cropping systems.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Li Q. Song X. Meng X. Zhang J. Zhang M. Chen L.Y. Li J. Wang B. Shaping future sugarcane: Ideal plant architecture and breeding strategies Mol. Plant 20251872572810.1016/j.molp.2025.04.00440205737 · doi ↗ · pubmed ↗
- 2Wen W. Gu S. Xiao B. Wang C. Wang J. Ma L. Wang Y. Lu X. Yu Z. Zhang Y. In situ evaluation of stalk lodging resistance for different maize (Zea mays L.) cultivars using a mobile wind machine Plant Methods 2019159610.1186/s 13007-019-0481-131452672 PMC 6701094 · doi ↗ · pubmed ↗
- 3Multani D.S. Briggs S.P. Chamberlin M.A. Blakeslee J.J. Murphy A.S. Johal G.S. Loss of an MDR transporter in compact stalks of maize br 2 and sorghum dw 3 mutants Science 2003302818410.1126/science.108607214526073 · doi ↗ · pubmed ↗
- 4Wei L. Zhang X. Zhang Z. Liu H. Lin Z. A new allele of the Brachytic 2 gene in maize can efficiently modify plant architecture Heredity 2018121758610.1038/s 41437-018-0056-329472693 PMC 5997708 · doi ↗ · pubmed ↗
- 5Zhang D. Sun W. Singh R. Zheng Y. Cao Z. Li M. Lunde C. Hake S. Zhang Z. GRF-interacting factor 1 Regulates Shoot Architecture and Meristem Determinacy in Maize Plant Cell 20183036037410.1105/tpc.17.0079129437990 PMC 5868708 · doi ↗ · pubmed ↗
- 6Tsuda K. Abraham-Juarez M.J. Maeno A. Dong Z. Aromdee D. Meeley R. Shiroishi T. Nonomura K.I. Hake S. KNOTTED 1 Cofactors, BLH 12 and BLH 14, Regulate Internode Patterning and Vein Anastomosis in Maize Plant Cell 2017291105111810.1105/tpc.16.0096728381444 PMC 5466031 · doi ↗ · pubmed ↗
- 7Depuydt S. Hardtke C.S. Hormone signalling crosstalk in plant growth regulation Curr. Biol.201121 R 365R 37310.1016/j.cub.2011.03.01321549959 · doi ↗ · pubmed ↗
- 8Zhao J. Yuan B. Zhang H. Guo X. Wang L. Qiu X. Xie Q. Mu L. Ma C. Zhou T. Phenotypic characterization and genetic mapping of the semi-dwarf mutant sdw 9 in maize Theor. Appl. Genet.202413725310.1007/s 00122-024-04762-239433551 · doi ↗ · pubmed ↗