Deciphering the Genetic Basis of Congenital Vertebral Malformations Through a Stepwise Diagnostic Approach
Anna Szoszkiewicz, Anna Sowińska-Seidler, Aleksandra Wnuk-Kłosińska, Ewelina Bukowska-Olech, Karolina Biel, Karolina Matuszewska, Marcin Biel, Magdalena Badura-Stronka, Renata Glazar, Anna Jakubiuk-Tomaszuk, Maciej Krawczyński, Krzysztof Szczałuba, Karolina Śledzińska

TL;DR
This study explores the genetic causes of congenital vertebral malformations using a stepwise genomic approach, identifying new genetic variants and candidate genes.
Contribution
The study expands the molecular spectrum of congenital vertebral malformations by identifying novel pathogenic variants and candidate genes.
Findings
A 12% diagnostic success rate was achieved using a three-tiered genomic approach in patients with congenital vertebral malformations.
Pathogenic variants were identified in genes such as FLNB and KMT2D, and a deletion upstream of SOX9 was detected.
Candidate genes NSD2 and TBXT were identified but require further functional validation.
Abstract
Congenital vertebral malformations (CVMs), affecting approximately 0.5–1 per 1000 live births, occur either in an isolated form or as part of syndromic disorders. Despite the identification of numerous causative genes for CVMs, the molecular etiology of most cases remains unknown. In this study, we applied a three-tiered diagnostic approach (chromosomal microarray analysis, followed by custom gene panel analysis, and exome/genome sequencing) in a cohort of 34 patients with CVMs. We achieved a 12% diagnostic success rate, identifying a deletion upstream of SOX9 and pathogenic or likely pathogenic variants in FLNB and KMT2D. Most pathogenic variants were detected by exome or genome sequencing, while earlier-tier analyses yielded limited results. We also identified two candidate genes, NSD2 and TBXT, that may contribute to the phenotype observed in our patients, but warrant future…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
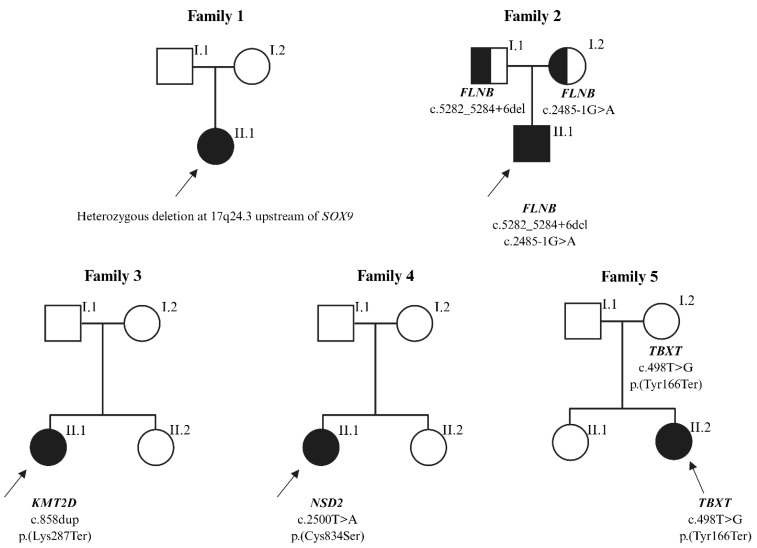 Figure 1
Figure 1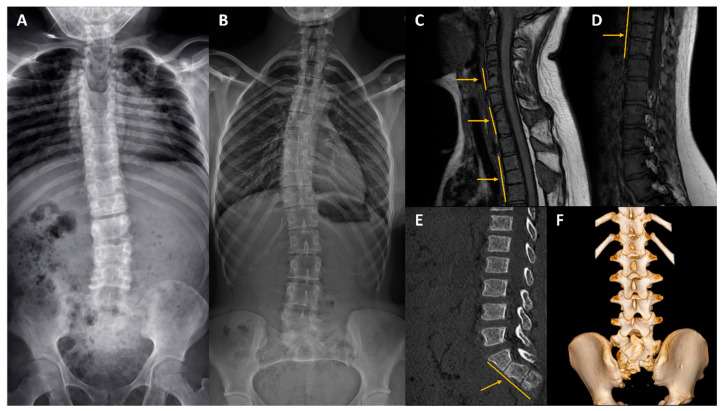 Figure 2
Figure 2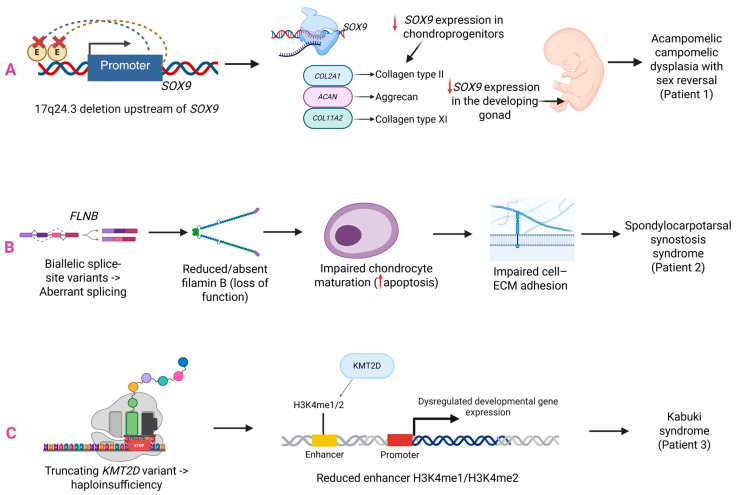 Figure 3
Figure 3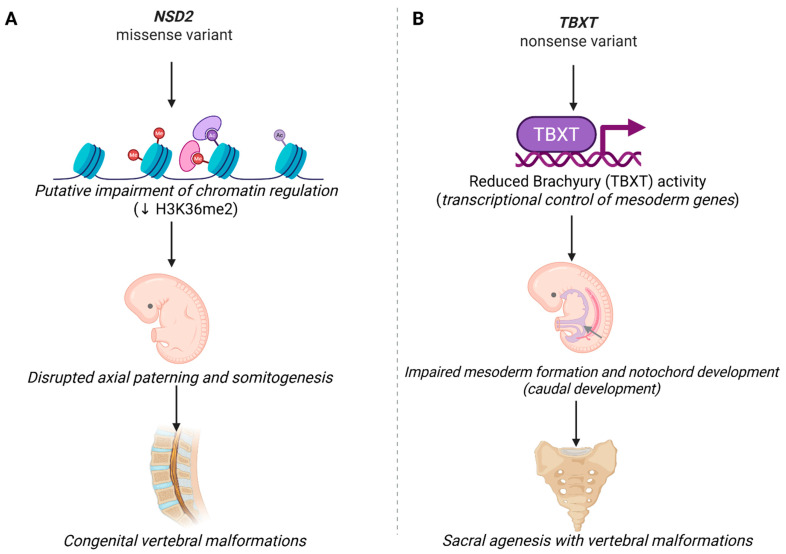 Figure 4
Figure 4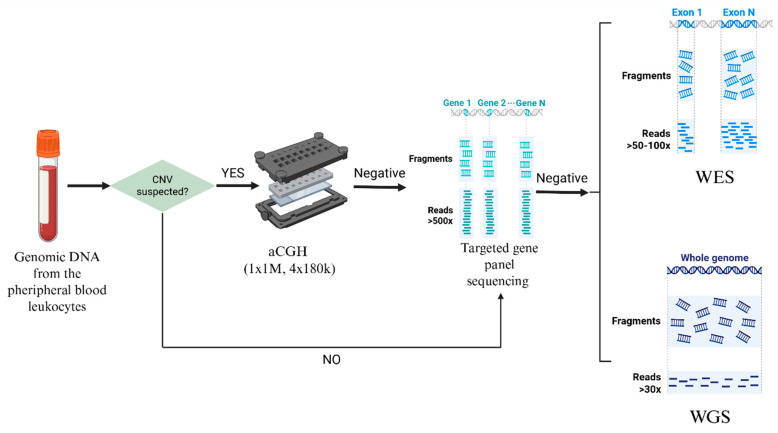 Figure 5
Figure 5- —Polish National Science Centre
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSpine and Intervertebral Disc Pathology · Connective tissue disorders research · Scoliosis diagnosis and treatment
1. Introduction
Congenital vertebral malformations (CVMs) are a heterogeneous group of skeletal disorders, causing chronic pain and disability. Their estimated prevalence is approximately 1–2 per 2000 live births, although the actual incidence is likely higher due to underdiagnosis. CVMs are classified as segmentation, formation, or mixed defects. The disorder may occur in isolation or as part of syndromes including Klippel–Feil, Alagille, Kabuki, CHARGE, and Spondylocarpotarsal synostosis syndromes [1,2,3,4,5]. Genetic factors explain only 10–20% of cases [6,7,8]. More than 100 genes have been linked to CVMs, most of which encode developmental signaling regulators crucial for spinal morphogenesis [4]. In addition, maternal drug intake and maternal diseases during pregnancy are environmental risk factors for CVMs.
Klippel–Feil syndrome (KFS) is a rare skeletal disorder characterized by the fusion of two or more cervical vertebrae. The classic clinical features of the disease are a short neck, a low posterior hairline, and limited neck motion. Other symptoms include scoliosis, Sprengel’s deformity, urinary or gastrointestinal malformations, congenital heart disease, hearing loss, and neurologic problems [9,10]. Pathogenic variants in GDF6, GDF3, MEOX1, MYO18B, and RIPPLY2 explain a subset of cases of KFS. In the last decade, next-generation (NGS) studies have identified several candidate genes [9,10,11,12,13]. Despite these advances, the etiology of most KFS cases is unresolved.
Although researchers have explained the molecular basis of specific syndromes associated with vertebral defects, the etiology of a substantial proportion of CVMs remains unclear. We investigated 34 patients with CVMs, including a notable subgroup presenting with KFS (n = 16). The genetic basis of the disease was examined using a tiered diagnostic strategy. The study represents the first systematic application of this approach in an international CVM cohort.
2. Results
2.1. Cohort Constitution
We summarized the phenotypic features of the patients in Table 1. We enrolled 34 probands diagnosed with CVM, primarily patients with severe scoliosis or kyphosis. We observed a slight female predominance, with a ratio of approximately 3:2. The affected patients ranged in age from 2 to 41 years. The most common vertebral anomalies were block vertebrae and hemivertebrae. KFS was the most frequent clinical and radiological diagnosis. CVMs occurred more often in the cervical and thoracic regions than in the lumbar, sacral, and coccygeal vertebrae. The most common extra-spinal anomalies were craniofacial dysmorphism (hypertelorism, epicanthus, micrognathia, facial asymmetry, low-set ears, and triangular face). Other consistent clinical features in our cohort included renal abnormalities (horseshoe or fused kidneys, unilateral renal agenesis, and pelvic malposition) and cardiovascular defects (atrial and ventricular septal defects and tricuspid valve insufficiency).
2.2. Genetic Results
Our diagnostic success was 12% (n = 4). Using array comparative genomic hybridization (aCGH), we detected a de novo interstitial deletion at 17q24.3 in a patient with acampomelic campomelic dysplasia. Breakpoint sequencing refined its size to 1.671 Mb (chr17:70,259,128–71,930,429; hg38). The deletion encompassed eleven known SOX9 upstream regulatory elements [14]. Targeted NGS of a custom 42-gene panel did not allow us to establish a molecular diagnosis in 12 patients with KFS. Consequently, 33 individuals proceeded to the next stage of the diagnostic workflow. Whole-exome sequencing (WES) revealed five heterozygous variants in the subsequent patients: c.2485-1G>A and c.5282_5284+6del in the FLNB gene (linked to spondylocarpotarsal synostosis syndrome), c.858dup p.(Lys287Ter) in the KMT2D gene (linked to Kabuki syndrome), c.2500T>A p.(Cys834Ser) in the NSD2 gene (linked to Rauch-Steindl syndrome), and c.498T>G p.(Tyr166Ter) in the TBXT gene (linked to sacral agenesis with vertebral anomalies). Among these, only the pathogenic variant in KMT2D has been previously reported in the medical literature. We summarized the genetic findings from our cohort in Table 2 and presented the segregation of the variants in Figure 1. The clinical characteristics of variant-positive patients are presented in Table 3, with representative imaging findings shown in Figure 2.
3. Discussion
The heterogeneity and complexity of the genetic architecture of CVMs pose a challenge to understanding the etiology of the disease. In this study, we reported 34 patients in whom we applied a three-step diagnostic protocol. The genetic analyses revealed a de novo 17q24.3 deletion upstream of SOX9, a pathogenic variant in KMT2D, two likely pathogenic variants in FLNB, and novel candidate variants in NSD2 and TBXT. The molecular diagnostic rate was 12%, aligning with yields from large cohort studies [15,16]. A detailed clinical and genetic description of Patient 1 has been published previously [14]. The remaining cases from our cohort are discussed below. WES provided a diagnosis in three patients, representing the most effective diagnostic method in our cohort. In patient 2, WES revealed compound heterozygous likely pathogenic splice-site variants in FLNB: c.2485-1G>A and c.5282_5284+6del, both of which were novel. The 2-year-old child exhibited clinical features of spondylocarpotarsal synostosis syndrome (SCT), including short stature, short neck, spinal lordosis, vertebral fusions, and dysmorphic facial features. Hand radiographs obtained at the time of evaluation did not demonstrate carpal synostosis. However, skeletal maturation was markedly delayed, with a left-hand bone age corresponding to 9 months for the long bones and 18 months for the carpal bones. Delayed carpal ossification has been reported in SCT, and carpal synostosis may not be detectable early in childhood, becoming apparent on follow-up [17]. Manifestations observed in fewer than 25% of individuals with SCT include brachydactyly, clinodactyly, clubfoot, cleft palate, and enamel hypoplasia [18,19,20]. The patient had clinodactyly of the fifth fingers but did not present with the other uncommon features. To date, approximately 40 cases of SCT have been documented, with only five patients having compound heterozygous variants in FLNB [21,22,23]. Most reported variants included SNVs, small indels, and intragenic deletions that result in nonsense or frameshifting effects [24,25]. Only one splice-site variant has been documented in a patient with SCT [21]. Our proband represents the first case of SCT caused by biallelic canonical splice-site variants in the FLNB gene. The detected variants are predicted to disrupt splicing and generate loss-of-function alleles. In two Flnb-deficient mouse models, homozygous loss of Flnb resulted in growth restriction and a pattern of skeletal abnormalities that resembled the FLNB-related recessive phenotype, including delayed endochondral ossification, reduced cartilage matrix, vertebral segmentation defects, vertebral and rib fusions, as well as abnormal spinal curvature (kyphosis/scoliosis). Experimental analyses indicated that loss of Flnb affected chondrocyte maturation, leading to increased apoptosis in developing skeletal elements (Figure 3B) [26,27]. Future studies should incorporate RNA sequencing to confirm the splicing defects of the variants and assess their functional consequences.
The second patient with an established molecular diagnosis (P3) carried a de novo nonsense variant in KMT2D (c.858dup, p.Lys287Ter), which confirmed the diagnosis of Kabuki syndrome (KS; OMIM #147920). Our proband showed the typical features of KS, including distinctive facial appearance, kidney defects, skeletal changes, and mild intellectual disability [28]. The same variant was reported in another individual with KS with a similar clinical picture [29]. Notably, the patient presented with a butterfly vertebra. CVMs have been reported only occasionally in KS and may extend the known skeletal phenotype associated with this condition [30,31]. The KMT2D gene encodes a histone methyltransferase involved in enhancer-associated H3K4 methylation. Disruption of this process can alter developmental transcription needed for normal skeletal development (Figure 3C). Studies in mice have shown that Kmt2d loss-of-function disrupts osteochondral differentiation and endochondral ossification, and recapitulates craniofacial and growth phenotypes seen in Kabuki syndrome [32,33]. Our case highlights the importance of considering KMT2D testing in patients with vertebral and multisystem developmental defects.
In addition to the confirmed disease-causing variants, our analysis identified novel candidate variants for CVMs. WES identified a de novo missense variant of uncertain significance (VUS) in NSD2 (c.2500T>A p.Cys834Ser) in patient 4 and a likely pathogenic variant in TBXT (c.498T>G, p.Tyr166Ter) in patient 5. A schematic summary of putative developmental mechanisms for NSD2 and TBXT is provided in Figure 4.
Patient 4 had a clinical phenotype highly concordant with Rauch-Steindl syndrome (RSS), i.e., microcephaly, facial dysmorphisms, intellectual disability, failure to thrive, short stature, and muscular hypotonia. The NSD2 gene encodes a histone lysine methyltransferase involved in chromatin regulation and orchestrates developmental gene-expression programs. Disruption of chromatin-mediated transcriptional regulation during early embryogenesis may interfere with somitogenesis and, consequently, compromise axial patterning and vertebral morphogenesis. While most pathogenic variants in the NSD2 gene are truncating, several de novo missense variants have been documented in individuals with RSS [34,35]. The strong phenotype concordance between the patient and the RSS phenotype sets, along with the de novo origin, suggests that the variant is a potential contributor to the disease. Further studies, including transcriptomic and methylation analyses, will be relevant to confirm our findings.
Patient 5 carried a likely pathogenic nonsense variant in TBXT (c.498T>G, p.Tyr166Ter). The variant was also present in a healthy mother of the proband. The TBXT gene encodes Brachyury, which plays a central role in mesoderm development and formation of the body axis. Pathogenic, mostly biallelic variants in TBXT have been reported in patients with sacral agenesis with vertebral anomalies [36,37,38]. CVMs observed in our patient are consistent with this clinical spectrum, indicating that TBXT may play a role in the disease mechanism. Although the disorder follows an autosomal recessive inheritance pattern, the proband carried a heterozygous loss-of-function variant inherited from her healthy mother. Similarly, another study described a heterozygous TBXT variant transmitted from an unaffected parent to a child with sacral agenesis [38]. Taking together, we suggest that the variant is more likely a strong predisposing factor than a single causative allele.
We assessed the identified genes using the STRING database. Overall, little direct functional connectivity was observed. A single association was noted between KMT2D and NSD2, reflecting their shared involvement in chromatin regulation. No broader interaction network was apparent among the remaining genes.
We were unable to obtain a definitive molecular diagnosis for most patients in our cohort, including all individuals with KFS. CVMs may result from complex genetic mechanisms, such as oligogenic, digenic, or polygenic inheritance. In addition, environmental and epigenetic changes can influence disease [4,9,39]. These factors, along with the limitations of current sequencing technologies, may explain why many patients still do not receive a genetic diagnosis. Future analyses should include RNA sequencing, methylation profiling, or long-read sequencing to improve the diagnostic process in unresolved cases.
Female patients were overrepresented in our cohort, including four of the five patients with a molecular finding. However, for FLNB, KMT2D, NSD2, SOX9, and TBXT, published cases include both sexes, and there is no clear evidence that variant effects on vertebral development are sex-dependent. The present study is underpowered to address sex effects, and this question should be revisited in larger cohorts.
In this study, we provide a combined clinical and molecular characterization of CVMs in a cohort of Polish patients. In addition to known disease-causing variants, we identified pathogenic or candidate variants in FLNB, KMT2D, NSD2, and TBXT, as well as a pathogenic deletion upstream of SOX9. Further progress in understanding CVM pathogenesis will require studies integrating genomic data with functional and epigenetic analyses.
4. Methods
4.1. Patient Recruitment and Clinical Evaluation
We enrolled 34 Polish patients with CVMs, who underwent physical examination and spinal imaging (radiography, computed tomography, and magnetic resonance imaging). Isolated and syndromic cases were eligible, including structural CVMs (e.g., hemivertebrae, butterfly vertebrae, and vertebral fusions) and KFS. We excluded patients with acquired vertebral deformities (post-traumatic, infectious, or neoplastic) as well as individuals with incomplete clinical data, no DNA available for testing, or a lack of written informed consent. We reviewed each participant’s medical records to define the vertebral malformation phenotype and identify any associated anomalies involving the spinal cord, heart, kidneys, brain, or other skeletal structures. This study was approved by the Institutional Review Board of the Poznan University of Medical Sciences ethics committee. Written informed consent for participation and publishing the information and images was obtained from all patients and the parents of underage participants before genetic testing.
4.2. Genetic Analyses
We extracted genomic DNA (gDNA) from peripheral blood leukocytes using the MagCore HF16 Automated Nucleic Acid Extractor (RBC Bioscience Corp., New Taipei City, Taiwan). The diagnostic workflow was implemented in a stepwise manner. First, aCGH was performed. Next, we applied targeted NGS of a custom gene panel to patients diagnosed with KFS. Finally, WES or whole-genome sequencing (WGS) was performed in cases that remained negative after the preceding analyses. All candidate genetic variants were further confirmed by segregation analysis using Sanger sequencing. Figure 5 depicts this study design and diagnostic workflow.
4.2.1. aCGH
Array comparative genomic hybridization (aCGH) was performed in patients with a clinical suspicion of pathogenic copy number variations (CNVs). Analyses were conducted using the SurePrint G3 Human CGH Microarray 1 × 1 M or 4 × 180 k platform (Agilent Technologies, Santa Clara, CA, USA) with a median probe spacing of 2.1 kb, according to the manufacturer’s instructions. Hybridization signals were recorded with the SureScan Dx Microarray Scanner (Agilent Technologies) and analyzed in Agilent CytoGenomics software (v5.0.2.5). The interpretation of CNVs drew on multiple reference resources, including DECIPHER, the Database of Genomic Variants (DGV), and Mouse Genome Informatics (MGI). Pathogenic CNVs were confirmed by quantitative PCR (qPCR) and evaluated for parental segregation. The qPCR procedure was performed as previously described [14].
4.2.2. Targeted NGS Panel
A cohort with KFS with negative aCGH results underwent targeted NGS. We designed a custom panel comprising 42 genes linked to CVMs using the Ion AmpliSeq Designer platform (v7.8.7; Thermo Fisher Scientific, Waltham, MA, USA) (Supplementary Table S1). We selected genes based on a review of the medical literature and their documented association with CVMs in genetic databases (OMIM and ClinVar). Libraries were prepared from 50 ng gDNA using the Ion AmpliSeq™ Library Kit 2.0 (Thermo Fisher Scientific, Waltham, MA, USA) and sequenced on the Ion Torrent S5 platform. The detailed laboratory workflow has been previously described [40]. Sequencing reads were processed using Torrent Suite v5.20.8.0 to perform base calling, quality control, and alignment to the GRCh37/hg19 reference genome. Variants were filtered using the following thresholds: read depth ≥ 20, PHRED quality ≥ 40, and allele frequency ≥ 0.15. NGS alignments were visualized using the Integrative Genomics Viewer (version 2.19.1; Broad Institute and the Regents of the University of California). For variant prioritization and interpretation, we evaluated the variants using reference databases, including HGMD, ClinVar, dbSNP, and gnomAD. The in silico prediction tools used in our analysis included SIFT, Polyphen-2, CADD, and REVEL. The variant’s pathogenicity was interpreted in accordance with the American College of Medical Genetics (ACMG) guidelines.
4.2.3. WES and WGS
WES or WGS was performed in certified diagnostic laboratories on DNA isolated from the patient’s blood samples. In WES, the coding regions and adjacent intronic sequences were enriched using a custom in-solution hybridization kit (Twist Bioscience, San Francisco, USA), achieving an average depth of approximately 50×. WGS libraries were prepared with the TruSeq DNA Nano Kit (Illumina, San Diego, CA, USA), yielding a mean depth of 30×. Prepared libraries were sequenced on the Illumina NovaSeq platform (Illumina, San Diego, USA). Sequencing reads were demultiplexed with bcl2fastq2, and adapters were removed using Skewer (v0.2.2). The reads were aligned to the GRCh38/hg38 human reference genome. PCR duplicates and low-quality reads were filtered out, and variant calling was performed using in-house bioinformatics software. CNV calling was performed for WGS as part of the diagnostic bioinformatics workflow, whereas WES analysis did not include CNV calling. First, we examined rare variants with a minor allele frequency (MAF) < 1%. In addition, pathogenic, likely pathogenic, or variants of uncertain significance in genes linked to CVMs were examined. Downstream analyses followed the workflow described for the targeted NGS panel.
4.2.4. Sanger Sequencing
We have analyzed the genomic region of interest using Sanger sequencing to confirm the presence of each pathogenic, likely pathogenic, and uncertain variant detected in NGS-based analyses. Primers were designed using the Primer3 tool version 0.4.0. PCR amplification and purification were performed according to standard laboratory protocols. Sanger sequencing was performed with dye-terminator chemistry (kit v.3, ABI 3130XL) and run on Applied Biosystems Prism 3700 DNA Analyzer.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chen Y. Liu Z. Chen J. Zuo Y. Liu S. Chen W. Liu G. Qiu G. Giampietro P.F. Wu N. The genetic landscape and clinical implications of vertebral anomalies in VACTERL association J. Med. Genet.20165343143710.1136/jmedgenet-2015-10355427084730 PMC 4941148 · doi ↗ · pubmed ↗
- 2Erol B. Tracy M.R. Dormans J.P. Zackai E.H. Maisenbacher M.K. O’Brien M.L. Turnpenny P.D. Kusumi K. Congenital scoliosis and vertebral malformations: Characterization of segmental defects for genetic analysis J. Pediatr. Orthop.20042467468210.1097/01241398-200411000-0001515502569 · doi ↗ · pubmed ↗
- 3Giampietro P.F. Dunwoodie S.L. Kusumi K. PourquiéO. Tassy O. Offiah A.C. Cornier A.S. Alman B.A. Blank R.D. Raggio C.L. Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans Ann. N. Y. Acad. Sci.20091151386710.1111/j.1749-6632.2008.03452.x 19154516 · doi ↗ · pubmed ↗
- 4Szoszkiewicz A. Bukowska-Olech E. Jamsheer A. Molecular landscape of congenital vertebral malformations: Recent discoveries and future directions Orphanet J. Rare Dis.2024193210.1186/s 13023-024-03040-038291488 PMC 10829358 · doi ↗ · pubmed ↗
- 5Turnpenny P.D. Alman B. Cornier A.S. Giampietro P.F. Offiah A. Tassy O. PourquiéO. Kusumi K. Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man Dev. Dyn.20072361456147410.1002/dvdy.2118217497699 · doi ↗ · pubmed ↗
- 6Liu J. Wu N. Yang N. Takeda K. Chen W. Li W. Du R. Liu S. Zhou Y. Zhang L. TBX 6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: Further evidence supporting the compound inheritance and TBX 6 gene dosage model Genet. Med.2019211548155810.1038/s 41436-018-0377-x 30636772 PMC 6659397 · doi ↗ · pubmed ↗
- 7Wu N. Ming X. Xiao J. Wu Z. Chen X. Shinawi M. Shen Y. Yu G. Liu J. Xie H. TBX 6 null variants and a common hypomorphic allele in congenital scoliosis N. Engl. J. Med.201537234135010.1056/NEJ Moa 140682925564734 PMC 4326244 · doi ↗ · pubmed ↗
- 8Zhao S. Zhang Y. Chen W. Li W. Wang S. Wang L. Zhao Y. Lin M. Ye Y. Lin J. Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS)J. Med. Genet.202158414710.1136/jmedgenet-2019-10682332381727 PMC 7802082 · doi ↗ · pubmed ↗