Integrated Prenatal Genetic Evaluation of Renal Agenesis: Chromosomal Microarray Analysis, Whole Exome Sequencing, and Outcome Correlations in 203 Fetuses
Na Zhang, Ruibin Huang, Fang Fu, Hang Zhou, Ru Li, Can Liao

TL;DR
This study examines the genetic causes and outcomes of fetal kidney absence in 203 cases, showing how different genetic tests help predict outcomes.
Contribution
The study clarifies the complementary roles of CMA and WES in prenatal evaluation of renal agenesis and identifies genetic patterns.
Findings
Chromosomal abnormalities were found in 7.4% of cases, including aneuploidies and pathogenic CNVs.
WES identified P/LP single nucleotide variants in 6.3% of cases, with higher yield in bilateral or non-isolated RA.
Postnatal follow-up confirmed RA in most liveborn cases, with some showing additional phenotypes.
Abstract
Objectives: To characterize the prenatal phenotypic spectrum, genetic findings, and pregnancy outcomes of fetal renal agenesis (RA), and to clarify the complementary roles of chromosomal microarray analysis (CMA) and whole exome sequencing (WES) in phenotype-stratified prenatal evaluation. Methods: This retrospective study included 203 RA fetuses between March 2017 and November 2025. All cases underwent genome-wide copy number variant (CNV) analysis, and selected cases underwent WES. Detection rates were compared across subgroups by laterality, isolated vs. non-isolated phenotype, fetal sex, and presence of extrarenal anomalies. Pregnancy outcomes and postnatal imaging follow-up were collected when available. A systematic literature review of prenatal genetic testing in RA fetuses was performed. Results: Among 203 fetuses, unilateral RA accounted for 92.6% of cases, and 65.0% were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1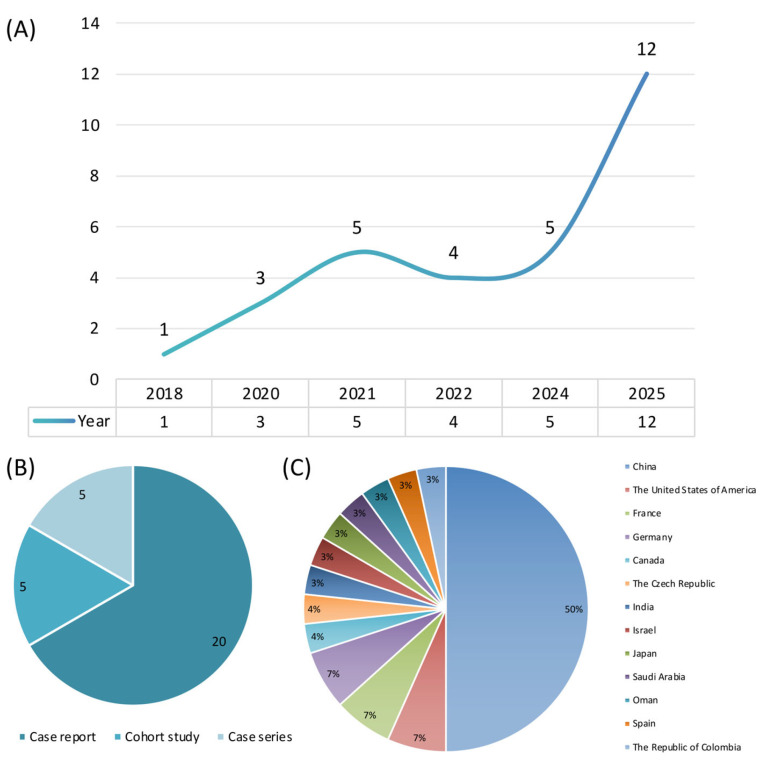 Figure 2
Figure 2- —Guangdong Basic and Applied Basic Research Foundation
- —National Natural Science Foundation of China
- —Guangzhou (Health) Science and Technology Project
- —Bureau of Science and Technology of Guangzhou, 2023 Municipal School (Institute) Enterprise Joint Funding Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPediatric Urology and Nephrology Studies · Prenatal Screening and Diagnostics · Fetal and Pediatric Neurological Disorders
1. Introduction
Renal agenesis (RA) is a severe developmental anomaly within the spectrum of congenital anomalies of the kidney and urinary tract (CAKUT), characterized by the complete absence of one or both kidneys due to disrupted nephrogenesis [1]. Population-based studies estimate the incidence of unilateral renal agenesis (URA) at approximately 1 in 1000–2000 births, whereas bilateral renal agenesis (BRA) is much rarer, occurring in approximately 1 in 3000–10,000 births [1,2,3]. The clinical consequences of RA are strongly dependent on laterality: URA is often compatible with long-term survival, while BRA is typically lethal because of severe oligohydramnios, pulmonary hypoplasia, and Potter sequence [1]. With the widespread use of second-trimester prenatal ultrasonography, RA is increasingly detected in utero, frequently in association with reduced amniotic fluid volume or additional structural anomalies [4]. Nevertheless, prenatal counseling remains particularly challenging, as fetal imaging alone often fails to reliably predict postnatal outcome, recurrence risk, and long-term prognosis.
Genetic heterogeneity represents a major contributor to the clinical complexity of RA. Both chromosomal abnormalities and monogenic disorders have been implicated in unilateral and bilateral forms [5,6,7,8,9,10,11,12,13,14]. Pathogenic copy number variants (CNVs), particularly recurrent microdeletions and microduplications involving 17p12 [11,12], 22q11.2 [5,6,9,12,15], and 16p11.2 [5,9,12], account for a proportion of cases, often through dosage-sensitive renal developmental genes such as HNF1B [16,17] and LHX1 [18]. Consequently, chromosomal microarray analysis (CMA) has become a first-tier genetic test for fetuses with CAKUT [19]. However, several studies have demonstrated that the diagnostic yield of CMA in isolated URA is relatively low, whereas its contribution is more substantial in non-isolated cases [5,6,12]. In contrast, whole exome sequencing (WES), particularly trio-based approaches, has emerged as a powerful tool for identifying monogenic causes of RA, especially in BRA, RA with extrarenal anomalies, or recurrent cases [7,8,10,13,14], revealing pathogenic variants in genes such as GREB1L [20,21,22], FRAS1 [10,14], FREM2 [14,23], NPNT [24], and GFRA1 [13,25]. Despite these advances, the optimal integration of CMA and WES in prenatal RA evaluation remains debated.
To date, most published studies have focused on selected subgroups of RA or applied heterogeneous testing strategies, limiting the direct comparison of diagnostic yields and their implications for prenatal counseling. In particular, data integrating detailed prenatal imaging, comprehensive genetic testing, and pregnancy outcomes within a single cohort remain scarce. In the present study, we retrospectively analyzed 203 RA fetuses at our center, systematically summarizing their prenatal imaging features, genetic testing results, and pregnancy outcomes. In parallel, we integrated evidence from previously published studies to evaluate the diagnostic performance of CMA and next-generation sequencing (NGS)-based testing across different RA subtypes. By combining institutional data with the existing literature, we aim to clarify the respective roles of CMA and WES in the prenatal evaluation of renal agenesis and to provide an evidence-based framework for prognostication and genetic counseling.
2. Materials and Methods
2.1. Study Design and Cohort
This retrospective study included 203 RA fetuses who were referred to the Prenatal Diagnosis Center of Guangzhou Women and Children’s Medical Center (GWCMC) between March 2017 and November 2025. The study protocol was approved by the Ethics Committee of GWCMC, and written informed consent was obtained from all participants. This retrospective cohort was conducted as part of the Specialist Alliance for Prevention and Control of Birth Defects (SAPCBD), which was initiated in 2021 by the Prenatal Diagnostic Center of GWCMC and consisted of 66 institutions in more than 10 provinces and has facilitated prenatal diagnostic evaluation in nearly 30,000 fetuses to date.
Clinical data were retrospectively collected from electronic medical records and included maternal and paternal age, gestational age at diagnosis, fetal sex, prenatal imaging findings, genetic testing results, and available pregnancy outcome information. Based on prenatal ultrasonographic findings, cases were classified as isolated or non-isolated RA. Isolated RA was defined as the presence of renal agenesis without additional congenital anomalies, whereas non-isolated RA was defined as renal agenesis accompanied by other anomalies, including additional CAKUT, soft markers, or extrarenal structural anomalies.
2.2. Genetic Testing and Variant Interpretation
Depending on gestational age and clinical indications, fetal samples were obtained from amniotic fluid, cord blood, or products of conception, while parental samples were collected from peripheral blood. Genomic DNA was extracted using standard protocols according to the manufacturer’s instructions after written informed consent was obtained.
All fetal samples initially underwent quantitative fluorescent polymerase chain reaction (QF-PCR) to exclude maternal cell contamination and to rapidly screen for common aneuploidies involving chromosomes 13, 18, 21, X, and Y. Genome-wide CNV analysis was subsequently performed using CMA in most cases, while CNV sequencing (CNV-seq) was applied in a small number of cases (n = 9). CMA was performed using the Affymetrix CytoScan HD or CytoScan 750K arrays (Affymetrix, Santa Clara, CA, USA), which integrate single-nucleotide polymorphism (SNP) array and array-based comparative genomic hybridization (aCGH) technologies, with an effective resolution of approximately 10 kb for deletions and 100 kb for duplications. CNV-seq was conducted using low-coverage whole genome sequencing to enable genome-wide detection of copy-number changes. All CNVs identified by either method were interpreted using the same clinical criteria and annotated according to the GRCh37/hg19 reference genome.
Trio-WES was performed in selected cases based on predefined clinical indications, including CNV analysis results that were negative or classified as variants of uncertain significance (VUS), sufficient DNA quantity and quality, and availability of parental samples. In addition, in some cases with advanced gestational age or strong parental preference, trio-WES was performed concurrently with CNV analysis as part of a one-step prenatal diagnostic approach. Targeted exome capture was carried out using the Agilent SureSelect Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA), followed by paired-end sequencing (150 bp reads) on Illumina HiSeq 2500, HiSeq X Ten, or NovaSeq platforms (Illumina, San Diego, CA, USA). Bioinformatic analysis of exome sequencing data was performed using an in-house pipeline, including sequence alignment, variant calling, quality control, variant filtration, and annotation. Variant interpretation incorporated inheritance patterns, fetal phenotypes, and family segregation analysis, with confirmation of candidate variants by Sanger sequencing when applicable. Sequence variants were classified according to the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines as pathogenic (P), likely pathogenic (LP), VUS, likely benign, or benign. For the purpose of diagnostic yield calculation, only P and LP variants were considered positive findings. Incidental findings (IFs) were reported in accordance with current ACMG recommendations. Prior to trio-WES, all families received pre-test genetic counseling, and written informed consent explicitly addressed the possibility of detecting incidental findings unrelated to the fetal renal phenotype, as well as the institutional reporting policy for such results. Detailed information regarding the analysis and interpretation of the WES data is provided in Supplementary File S1.
2.3. Statistical Analysis
Statistical analyses were performed using SPSS software (version 26.0; IBM Corp., Armonk, NY, USA). Categorical variables, including detection rates across clinical subgroups stratified by laterality (URA vs. BRA), fetal sex, isolated versus non-isolated renal agenesis, and the presence or absence of extrarenal anomalies, were summarized as counts and percentages and compared using Fisher’s exact test. Continuous variables, including maternal age, paternal age, and gestational age at diagnosis, were expressed as mean ± standard deviation (SD) and compared between groups using Welch’s t-test. All statistical tests were two-sided, and p-value < 0.05 was considered statistically significant.
3. Results
3.1. Clinical Characteristics and Prenatal Imaging Features of RA Fetuses
A total of 203 fetuses diagnosed with RA were included in this study. All cases were referred to the Prenatal Diagnosis Center of GWCMC between March 2017 and November 2025. The mean maternal age at diagnosis was 30.14 ± 4.06 years, and the mean paternal age was 32.34 ± 4.98 years. The mean gestational age at diagnosis was 24.72 ± 3.18 weeks. Among the affected fetuses, 77 (37.9%, 77/203) were male and 126 (62.1%, 126/203) were female. Most cases were diagnosed during the second trimester and underwent amniocentesis, accounting for 172 fetuses (84.7%, 172/203). Twenty-two cases (10.8%, 22/203) were diagnosed at a later gestational age and underwent percutaneous umbilical blood sampling. The remaining nine cases (4.4%, 9/203) were diagnosed in the context of severe oligohydramnios or anhydramnios and underwent genetic testing using products of conception. Notably, all nine cases with pregnancy tissue samples showed bilateral renal involvement. RA predominantly presented as unilateral involvement, accounting for 188 cases (92.6%, 188/203), whereas bilateral renal agenesis was observed in 15 cases (7.4%, 15/203). Among unilateral cases, left-sided RA was identified in 86 (42.4%, 86/203) fetuses and right-sided RA in 102 (50.2%, 102/203) fetuses. When RA and concomitant CAKUT were considered together, renal involvement was unilateral in 172 (84.7%, 172/203) fetuses and bilateral in 31 (15.3%, 31/203) fetuses.
Based on prenatal ultrasonographic findings, 132 (65.0%, 132/203) cases were classified as isolated, while 71 (35.0%, 71/203) cases were classified as non-isolated, showing additional CAKUT features, soft markers, or extrarenal structural anomalies. A total of 17 (8.4%, 17/203) fetuses exhibited additional CAKUT phenotypes, including contralateral duplicated kidney or duplex collecting system (n = 5), compensatory enlargement of the contralateral kidney (n = 2), contralateral renal dysplasia (n = 5), contralateral hydronephrosis or pyelectasis (n = 3), renal cysts (n = 1), and ectopic kidney (n = 1). Extrarenal structural anomalies were identified in 46 (22.7%, 46/203) fetuses. Cardiovascular anomalies were the most frequent, affecting 14 cases, including ventricular septal defects in 8 fetuses. Other extrarenal anomalies included multisystem anomalies (n = 10), skeletal anomalies (n = 8), fetal growth restriction (n = 8), central nervous system anomalies (n = 3), gastrointestinal anomalies (n = 2), and respiratory system anomalies (n = 1). Soft markers were detected in 36 (17.7%, 36/203) fetuses, with single umbilical artery being the most common finding, present in 20 cases. Oligohydramnios or anhydramnios was observed in 20 (9.9%, 20/203) fetuses, including 15 BRA cases, four URA cases combined with contralateral renal abnormalities, and one case with isolated left RA associated with multiple extrarenal anomalies.
3.2. Diagnostic Yield and Spectrum of Chromosomal Abnormalities
CMA was performed in all 203 fetuses with renal agenesis. Clinically significant chromosomal abnormalities were identified in 15 cases, yielding an overall diagnostic rate of 7.4% (15/203). VUS were detected in four additional cases (2.0%, 4/203). Among the 15 fetuses with clinically significant chromosomal abnormalities detected by CMA, four cases were diagnosed with whole-chromosome aneuploidies, including trisomy 18 (n = 1), 47, XXY (n = 1), 47, XXX (n = 1), and mosaic 47, XXX (n = 1). The remaining 11 cases harbored P/LP CNVs beyond whole-chromosome aneuploidies, including nine deletions and two duplications. Six of the 15 fetuses presented with isolated RA, whereas the remaining nine cases were classified as non-isolated RA and were associated with additional CAKUT features, extrarenal anomalies, or both. Parental studies were available for nine cases, confirming six de novo abnormalities and three inherited abnormalities; inheritance could not be determined in the remaining cases. Detailed genomic coordinates, CNV sizes, inheritance patterns, associated phenotypes, and pregnancy outcomes are summarized in Table 1.
Stratified analyses according to laterality, fetal sex, the presence of associated anomalies, and the presence of extrarenal anomalies revealed no statistically significant differences in the detection rate of clinically significant chromosomal abnormalities. Specifically, the detection rates were 7.4% (14/188) in URA and 6.7% (1/15) in BRA cases (p = 1.000), and 10.4% (8/77) in male fetuses versus 5.6% (7/126) in female fetuses (p = 0.269). The detection rate was 5.3% (7/132) in isolated and 11.3% (8/71) in non-isolated cases (p = 0.159), and was higher in fetuses with extrarenal anomalies compared with those without extrarenal anomalies [11.1% (7/63) vs. 5.7% (8/140)], although the difference was not statistically significant (p = 0.244). Comparisons of continuous clinical variables showed no significant differences between CMA-positive and CMA-negative groups, including maternal age (30.2 ± 3.3 vs. 30.1 ± 4.1 years, p = 0.913), paternal age (31.5 ± 4.1 vs. 32.4 ± 4.9 years, p = 0.431), or gestational age at diagnosis (25.7 ± 3.7 vs. 24.6 ± 3.1 weeks, p = 0.279).
3.3. Diagnostic Yield and Molecular Spectrum of Trio-WES
Trio-WES was subsequently performed in 127 RA fetuses, and eight P/LP single nucleotide variants (SNVs) were identified, yielding an overall diagnostic rate of 6.3% (8/127). When fetuses carrying LP/VUS compound heterozygous variants were additionally included, a total of 11 fetuses (8.7%, 11/127) were found to harbor SNVs with potential clinical relevance (defined as P/LP and LP/VUS variants). In addition to these findings, VUS were identified in eight fetuses (6.3%, 8/127). IFs unrelated to the fetal renal phenotype were detected in three cases (2.4%, 3/127), including two fetuses with pathogenic GJB2 variants associated with hearing loss and one fetus with a pathogenic PKP2 variant associated with arrhythmogenic cardiomyopathy (Table 2). Stratified analyses revealed significant differences in detection rates across clinical subgroups. The detection rate was significantly higher in BRA fetuses than in those with URA [30.8% (4/13) vs. 6.1% (7/114), p = 0.015]. Similarly, non-isolated was associated with a higher detection rate compared with isolated cases [15.8% (9/57) vs. 2.9% (2/70), p = 0.012], and fetuses with extrarenal anomalies showed a markedly higher yield than those without such abnormalities [18.4% (9/49) vs. 2.6% (2/78), p = 0.003]. In contrast, no significant difference in detection rate was observed between male and female fetuses [4.4% (2/45) vs. 11.0% (9/82), p = 0.326]. Comparisons of continuous variables showed no statistically significant differences in maternal age (29.2 ± 3.6 vs. 29.8 ± 4.0 years, p = 0.610) or paternal age (32.5 ± 3.4 vs. 32.1 ± 4.9 years, p = 0.727) between SNV-positive and SNV-negative cases. The gestational age at diagnosis tended to be lower in SNV-positive cases than in SNV-negative cases (22.5 ± 3.1 vs. 24.5 ± 3.3 weeks), although this difference did not reach statistical significance (p = 0.064).
Among the 11 fetuses harboring SNVs with potential clinical relevance, three carried compound heterozygous variants in FRAS1, which was the only gene identified in more than one case, whereas the remaining eight fetuses each harbored variants in distinct disease-associated genes, including EYA1, KMT2A, FGFR2, PBX1, PUF60, KMT2D, FANCI, and LTBP3. With respect to inheritance patterns, six cases were consistent with autosomal dominant inheritance, including five de novo variants and one variant inherited from the father, while the remaining five cases followed an autosomal recessive pattern, all of which carried compound heterozygous variants inherited from both parents. A total of 13 P/LP SNVs were identified in these cases. Variant classification revealed a predominance of loss-of-function variants, including five nonsense variants, three frameshift variants, and three canonical splice-site variants, accounting for the majority of pathogenic findings. In addition, one missense variant and one in-frame deletion were identified.
3.4. Pregnancy Outcomes and Postnatal Follow-Up
Among the 203 RA fetuses, pregnancy outcomes were successfully obtained for 149 cases, while 56 cases were lost to follow-up or lacked postnatal reassessment. A total of 59 pregnancies were terminated. Among these, 22 fetuses carried P/LP CNVs or SNVs, 18 were associated with additional extrarenal structural anomalies, six presented with BRA, two had URA combined with contralateral renal abnormalities and oligohydramnios, and one fetus carried a VUS SNV. The remaining ten pregnancies were terminated for other reasons. Eighty-eight fetuses were delivered and subsequently underwent postnatal imaging follow-up. Postnatal imaging confirmed the diagnosis of RA in 84 cases, among which compensatory hypertrophy of the contralateral kidney was observed in nine cases. In four cases, pelvic ectopic kidneys were diagnosed postnatally; none of these fetuses had undergone prenatal fetal magnetic resonance imaging (MRI). During postnatal follow-up, seven children were found to have newly identified clinical phenotypes that had not been detected prenatally, including scoliosis, sacrococcygeal teratoma, psychomotor developmental delay, ventricular septal defect, subependymal hemorrhage, cleft palate, and intracranial hemorrhage. In five of these cases, trio-WES had been performed prenatally with negative results, and reanalysis of the sequencing data after incorporating newly identified postnatal phenotypes remained negative.
3.5. Summary of Published Evidence on Prenatal Genetic Testing in RA
A systematic literature search and review identified a total of 30 studies reporting prenatal genetic testing in RA fetuses that were included for further analysis (Figure 1, Supplementary File S2). The included publications comprised five cohort studies [5,6,7,10,12], five case series [8,9,11,13,14], and twenty case reports [15,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. With respect to publication year, one study was published in 2018, three in 2020, five in 2021, four in 2022, five in 2024, and twelve in 2025 (Figure 2). Geographically, the studies originated predominantly from China (n = 15), followed by the United States (n = 2), France (n = 2), Germany (n = 2), Canada (n = 1), the Czech Republic (n = 1), India (n = 1), Israel (n = 1), Japan (n = 1), Saudi Arabia (n = 1), Oman (n = 1), Spain (n = 1), and the Republic of Colombia (n = 1). Regarding genetic testing strategies, CMA was applied in six studies [5,6,9,11,12,15], targeted gene panels in two studies [10,23], genome sequencing (GS) in two studies [13,29], and WES in twenty studies [7,8,14,20,21,22,24,25,26,27,28,30,31,32,33,34,35,36,37,38]. Among the five cohort studies, three employed CMA and reported diagnostic yields ranging from 2.47% to 17.19%, with a pooled yield of 10.34% (24/232). One cohort study using a targeted gene panel reported a diagnostic yield of 15.38% (6/39), and one cohort study applying WES reported a diagnostic yield of 16.67% (2/12).
Across the 30 included studies, a total of 71 RA fetuses were reported to harbor P/LP genetic findings. Of these, 32 cases were identified by CMA, with the most frequently reported chromosomal abnormalities being 16p11.2 microdeletion (n = 7) and 22q11.21 microdeletion (n = 7). The remaining 39 cases were detected by NGS, most commonly involving variants in FRAS1 (n = 8), followed by FREM2 (n = 3), GFRA1 (n = 3), GREB1L (n = 3), PAX2 (n = 2), and SALL4 (n = 2). Among the 71 genetically positive fetuses with RA reported in the literature, 35 presented with isolated renal agenesis. Of these, 19 cases (54.3%) were attributed to CNVs. Recurrent CNVs predominantly involved 22q11.21 microdeletion (5/35, 14.3%) and 16p11.2 copy-number alterations (5/35, 14.3%), including four microdeletions and one microduplication, followed by 17p12 microdeletion (2/35, 5.7%), whereas all other CNVs were reported only once. The remaining 16 cases (45.7%) harbored sequence-level variants identified by NGS, with the most frequently implicated genes being GFRA1 (3/35, 8.6%) and GREB1L (3/35, 8.6%), followed by FRAS1 (2/35, 5.7%) and FREM2 (2/35, 5.7%); all other genes were reported in single cases.
4. Discussion
4.1. Principal Findings and Clinical Relevance
In this study, we systematically evaluated the prenatal phenotype, genetic findings, and pregnancy outcomes of a large cohort of 203 RA fetuses and further integrated evidence from previously published studies. Using a combined strategy of genome-wide CNV analysis and trio-WES, we demonstrated that clinically significant genetic abnormalities are identifiable in a substantial proportion of RA cases, with distinct diagnostic yields across different clinical subgroups. Chromosomal abnormalities accounted for 7.4% of cases, whereas trio-WES provided an additional diagnostic yield of 6.3% based on P/LP variants, increasing to 8.7% when variants with potential clinical relevance were considered. Notably, the diagnostic yield of WES was significantly enriched in BRA fetuses, non-isolated phenotypes, and extrarenal structural anomalies. These findings highlight the pronounced genetic heterogeneity underlying RA and underscore the clinical value of a phenotype-driven, stepwise genetic testing strategy for optimizing prenatal counseling and risk assessment.
4.2. Prenatal Phenotype Spectrum and Counseling Challenges
Renal agenesis exhibited marked phenotypic heterogeneity in our cohort, posing substantial challenges for prenatal counseling. Although unilateral renal agenesis predominated, more than one-third of fetuses presented with non-isolated phenotypes, including additional CAKUT features, soft markers, or extrarenal structural anomalies, most frequently involving the cardiovascular system. In addition, oligohydramnios or anhydramnios was almost exclusively observed in fetuses with BRA or URA accompanied by contralateral renal abnormalities, underscoring the close relationship between laterality, renal functional reserve, and amniotic fluid volume. These findings are consistent with previous reports indicating that the prenatal presentation of RA spans a broad clinical spectrum and that sonographic features alone are often insufficient to accurately predict postnatal outcome.
Importantly, our postnatal follow-up further highlighted the limitations of prenatal imaging. Pelvic ectopic kidneys were identified after birth in four fetuses initially diagnosed with RA, none of whom had undergone fetal MRI, suggesting that renal malposition may be misclassified as agenesis in the prenatal setting. Moreover, additional clinical phenotypes emerged postnatally in a subset of children, several of whom had undergone prenatal trio-WES with negative results and remained negative after reanalysis incorporating newly identified features. Together, these observations emphasize that prenatal assessment of RA is inherently constrained by incomplete phenotypic information and evolving postnatal manifestations, reinforcing the need for integrated genetic testing strategies and longitudinal follow-up to improve prognostic accuracy and counseling.
4.3. Chromosomal Abnormalities and the Role of CNV Analysis in Fetal RA
In our cohort, clinically significant chromosomal abnormalities were identified in 7.4% of fetuses with renal agenesis by genome-wide CNV analysis, supporting the role of CNV testing as a foundational component of prenatal genetic evaluation in RA. This diagnostic yield falls within the range reported by previous cohort studies, in which CMA-based detection rates varied from approximately 2% to 17%, with an overall yield of around 10% [5,6,12]. Consistent with prior reports, the majority of pathogenic CNVs identified in our study were deletions rather than duplications, and recurrent regions such as 16p11.2 and 22q11.21 were among the most frequently implicated loci in the published literature, underscoring their relevance to renal development. Despite these findings, CNV analysis demonstrated limited discriminatory power across clinical subgroups in our cohort. Although higher detection rates were observed in non-isolated cases and in fetuses with extrarenal anomalies, these differences did not reach statistical significance, likely reflecting the relatively small number of CNV-positive cases and the genetic heterogeneity of RA. These negative subgroup comparisons should be interpreted cautiously given limited statistical power. Therefore, larger multicenter cohorts and/or meta-analyses will be necessary to robustly evaluate subgroup-specific CMA yields and to determine whether clinically meaningful differences exist across phenotypic strata. Importantly, a subset of isolated RA cases also harbored pathogenic chromosomal abnormalities, indicating that the absence of additional sonographic anomalies does not preclude an underlying chromosomal etiology. Taken together, these observations suggest that CNV analysis should be routinely considered in the prenatal evaluation of RA, irrespective of phenotypic complexity, while acknowledging that its diagnostic yield alone may be insufficient to fully elucidate the genetic basis of the condition.
4.4. Added Value of Trio-WES and Gene-Disease Correlations in Fetal RA
Our study further confirms the incremental diagnostic value of trio-WES in the prenatal evaluation of RA, particularly when applied in a phenotype-stratified manner. Although the overall diagnostic yield of trio-WES based on P/LP variants was relatively modest (6.3%), the detection rate increased significantly in specific clinical subgroups, including fetuses with BRA, non-isolated RA, and those with extrarenal structural anomalies. In fetuses with BRA, the diagnostic yield approached one third, indicating that monogenic etiologies play a particularly prominent role in severe or syndromic forms of RA. These findings are highly consistent with previous reports, in which exome sequencing was preferentially applied to selected subgroups of fetuses with RA and yielded diagnostic rates typically ranging from 15% to 17% [7,10].
In our cohort, sequence-level variants in KMT2A and FRAS1 were identified in fetuses with isolated RA. When integrated with previously published studies, isolated RA has been associated with a diverse set of disease-associated genes, including KMT2A, FRAS1 [14], FREM1 [37], FREM2 [14], GFRA1 [13,25], GREB1L [20,21,22], PAX2 [28], NPNT [24], EYA1 [10], HOXC4 [8], and PRPF8 [8] (Table 3). Although each of these genes has been reported in only a limited number of cases, they converge on key biological pathways essential for kidney development, supporting the presence of genuine monogenic etiologies even in apparently isolated presentations. These genes play critical roles in early renal and urinary tract development, including ureteric bud induction, metanephric mesenchyme differentiation, extracellular matrix homeostasis, and epithelial–mesenchymal interactions. Disruption of these pathways may impair ureteric bud formation or reciprocal signaling, ultimately resulting in renal developmental failure or complete agenesis.
Of particular note, FRAS1, FREM1 and FREM2 encode components of the Fraser protein complex, which is essential for maintaining epithelial–mesenchymal structural integrity during embryogenesis [39]. Pathogenic variants in these genes cause Fraser syndrome, a multisystem developmental disorder characterized in both humans and animal models by cryptophthalmos, syndactyly, and genitourinary anomalies, among which RA represents a common and clinically important feature [40]. Experimental studies in animal models have demonstrated that loss of function of the Fraser complex disrupts basement membrane integrity and interferes with organ morphogenesis, providing a clear mechanistic explanation for renal and urinary tract involvement [41,42,43]. The identification of variants in these genes in RA fetuses in our study, even in the absence of overt syndromic features at the time of prenatal assessment, suggests that RA may represent an early or incomplete manifestation within a broader spectrum of developmental disorders.
Taken together, these findings indicate that isolated RA should not be regarded as a genetically “low-risk” phenotype. Instead, selective application of WES may be justified in carefully phenotyped isolated cases, particularly when combined with detailed prenatal assessment and longitudinal postnatal follow-up.
4.5. Pregnancy Outcomes and Implications for Prenatal Counseling
Pregnancy outcomes in our cohort further underscore the importance of integrated prenatal assessment in the management of RA fetuses. Termination of pregnancy was more frequently observed in cases with severe phenotypes, including BRA, oligohydramnios or anhydramnios, the presence of extrarenal structural anomalies, and the identification of P/LP genetic variants. This pattern is consistent with current clinical practice, in which the severity of fetal structural abnormalities and their potential genetic etiology substantially influence parental decision-making.
Among fetuses that were delivered and successfully followed postnatally, the diagnosis of RA was confirmed in most cases, and compensatory hypertrophy of the contralateral kidney was observed in a subset of children with URA, indicating favorable physiological adaptation. It should be noted that a proportion of cases were lost to follow-up or lacked postnatal reassessment, which is common in real-world practice at tertiary referral centers, as many families deliver and continue pediatric care at local hospitals, relocate, or are unable/unwilling to return for follow-up visits. This incomplete follow-up may introduce bias in outcome estimation and could lead to under-recognition of later-onset or milder postnatal phenotypes. However, postnatal follow-up also revealed newly identified clinical phenotypes that had not been detected prenatally, including neurodevelopmental abnormalities, congenital heart defects, and anomalies involving other organ systems. Notably, in several of these cases, prenatal trio-WES had yielded negative results, and reanalysis of the sequencing data after incorporating newly identified postnatal phenotypes remained negative. This may reflect both technical and interpretative limitations of exome-based testing, including reduced sensitivity for non-coding or deep intronic variants, low-level mosaicism, and certain structural rearrangements; moreover, even with updated phenotypic information, the available evidence supporting some gene-disease associations or variant-level pathogenicity may remain insufficient to enable definitive classification, particularly for rare or novel variants. In addition, incomplete penetrance, variable expressivity, and potential oligogenic/multifactorial mechanisms may further increase the complexity of genotype–phenotype matching. These findings highlight the current technical and interpretative limitations of prenatal genetic testing and reflect the dynamic and evolving nature of genotype–phenotype correlations.
Beyond the diagnostic yield for RA, trio-WES may also reveal IFs unrelated to the fetal renal phenotype but potentially relevant to postnatal care. In our cohort, IFs were identified in 2.4% (3/127) of RA fetuses, including two cases with pathogenic homozygous GJB2 variants associated with hearing loss and one case with a pathogenic heterozygous PKP2 variant associated with arrhythmogenic cardiomyopathy. Although these results did not explain RA, they may still inform postnatal management, such as early audiological evaluation and timely cardiology assessment when appropriate. Therefore, pre-test counseling and written informed consent should explicitly address the possibility of non-kidney diagnoses and clarify the institutional reporting policy for such findings, together with plans for post-test genetic counseling and follow-up.
From a clinical management perspective, our findings support a tiered, phenotype-driven genetic testing strategy for RA fetuses. CMA should remain the first-tier test for the detection of chromosomal abnormalities, whereas trio-WES can substantially increase the diagnostic yield in selected cases, particularly those with BRA, non-isolated phenotypes, or associated extrarenal structural anomalies. However, in time-sensitive prenatal diagnostic settings, a strictly sequential workflow may be constrained by gestational age and test turnaround time; it is generally feasible in early pregnancy and the early second trimester. For late-presenting cases-especially those beyond 25 weeks of gestation-we recommend a “one-step” approach, performing CNV analysis and trio-WES concurrently, to shorten time-to-results and support timely decision-making. At the same time, the persistence of unresolved cases despite the emergence of additional postnatal phenotypes underscores the importance of long-term follow-up and periodic data reanalysis, rather than the premature exclusion of a genetic etiology. Accordingly, prenatal counseling for renal agenesis should not rely solely on isolated imaging or genetic findings but should integrate structural severity, genetic testing results, and postnatal follow-up information. Such an approach enables more refined risk stratification, avoids overly deterministic prognostic conclusions, and provides families with more comprehensive and individualized decision-making support.
4.6. Strengths and Limitations
The major strengths of this study include the large cohort from a tertiary referral center with a standardized clinical workflow, comprehensive genetic testing, and pregnancy outcome data, and the parallel synthesis of institutional findings with published evidence. The single-center design enabled consistent phenotyping, uniform laboratory procedures, and harmonized variant interpretation across the entire cohort. By applying both CMA and trio-WES in a phenotype-stratified manner, we were able to directly compare their diagnostic performance across different subtypes of RA and to delineate clinical scenarios in which each modality provides the greatest utility. Importantly, the inclusion of postnatal follow-up and reanalysis further enhances the clinical relevance of our findings and reflects real-world prenatal diagnostic practice.
Several limitations should be acknowledged. First, this was a retrospective study conducted at a single tertiary referral center, which may introduce referral bias toward more complex or severe phenotypes, and may limit the generalizability of our diagnostic yields to broader prenatal populations. In particular, the case mix in a tertiary setting may be enriched for fetuses with more severe renal phenotypes, extrarenal anomalies, or families seeking advanced genetic testing, potentially inflating the observed yield compared with community-based cohorts. Second, although trio-WES substantially improved diagnostic yield in selected subgroups, a proportion of cases with evolving postnatal phenotypes remained genetically unresolved even after reanalysis, reflecting both the intrinsic blind spots of exome-based testing and current constraints in variant interpretation and gene-disease evidence. Third, functional validation was not performed in this study, which may limit confirmation of variant pathogenicity and inference of disease mechanisms, particularly for novel variants or VUS. However, functional studies are often more challenging to implement in the prenatal setting due to limited access to disease-relevant tissue samples and the narrow time window for clinical decision-making.
5. Conclusions
In conclusion, our study demonstrates that renal agenesis exhibits marked genetic heterogeneity and that the diagnostic yield of prenatal genetic testing is highly dependent on clinical context. CMA remains an essential first-tier test, whereas trio-WES provides significant incremental value in fetuses with bilateral involvement, non-isolated phenotypes, or extrarenal anomalies. Isolated RA, although associated with a lower diagnostic yield, may still harbor monogenic etiologies and should not be considered genetically benign. An integrated, phenotype-driven diagnostic strategy combined with longitudinal follow-up and data reanalysis offers a rational framework for improving prenatal counseling, refining prognostic assessment, and guiding clinical decision-making in families affected by fetal renal agenesis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jelin A. Renal agenesis Am. J. Obstet. Gynecol.2021225 B 28B 3010.1016/j.ajog.2021.06.04834507792 · doi ↗ · pubmed ↗
- 2Cai M. Lin N. Su L. Wu X. Xie X. Li Y. Chen X. Dai Y. Lin Y. Huang H. Detection of copy number disorders associated with congenital anomalies of the kidney and urinary tract in fetuses via single nucleotide polymorphism arrays J. Clin. Lab. Anal.202034 e 2302510.1002/jcla.2302531506986 PMC 6977156 · doi ↗ · pubmed ↗
- 3Delmas H.L. Kohler M. Doray B. Lemery D. Francannet C. Quistrebert J. Marie C. Perthus I. Congenital unilateral renal agenesis: Prevalence, prenatal diagnosis, associated anomalies. Data from two birth-defect registries Birth Defects Res.20171091204121110.1002/bdr 2.106528722320 · doi ↗ · pubmed ↗
- 4Society for Maternal-Fetal Medicine Norton M.E. Cheng Y. Chetty S. Chyu J.K. Connolly K. Ghaffari N. Hopkins L.M. Jelin A. Mardy A. SMFM Fetal Anomalies Consult Series #4: Genitourinary anomalies Am. J. Obstet. Gynecol.2021225 B 2B 3510.1016/j.ajog.2021.06.00934507800 PMC 8763622 · doi ↗ · pubmed ↗
- 5Su J. Qin Z. Fu H. Luo J. Huang Y. Huang P. Zhang S. Liu T. Lu W. Li W. Association of prenatal renal ultrasound abnormalities with pathogenic copy number variants in a large Chinese cohort Ultrasound Obstet. Gynecol.20225922623310.1002/uog.2370234090309 · doi ↗ · pubmed ↗
- 6Sagi-Dain L. Maya I. Peleg A. Reches A. Banne E. Baris H.N. Tenne T. Singer A. Ben-Shachar S. Microarray analysis in pregnancies with isolated unilateral kidney agenesis Pediatr. Res.20188382582810.1038/pr.2018.329320483 · doi ↗ · pubmed ↗
- 7Qin Y. Wang B. Zhu Y. Liu L. Liu N. Yao Y. Li H. Xu R. Zhang C. Song J. Prenatal diagnosis of fetuses with renal abnormalities: A retrospective analysis of 329 Chinese cases Orphanet J. Rare Dis.20252048610.1186/s 13023-025-04001-x 40993696 PMC 12462046 · doi ↗ · pubmed ↗
- 8Merz L.M. Kolvenbach C.M. Wang C. Mertens N.D. Seltzsam S. Mansour B. Zheng B. Schneider S. Schierbaum L. Hölzel S. Trio exome sequencing identifies de novo variants in novel candidate genes in 19.62% of CAKUT families Genet. Med.20252710143210.1016/j.gim.2025.10143240223730 PMC 12229778 · doi ↗ · pubmed ↗