Multi-State Structural Genomics Enables Large-Scale, Mechanistic, and Context-Specific Classification of ABCC6 Genetic Variants Implicated in Calcification Diseases
Jessica B. Wagenknecht, Neshatul Haque, Salomao D. Jorge, Brian D. Ratnasinghe, Raul Urrutia, William A. Gahl, Shira G. Ziegler, Michael T. Zimmermann

TL;DR
This paper uses 3D protein modeling to better understand how genetic changes in ABCC6 cause calcification diseases, helping classify uncertain variants.
Contribution
The study introduces a novel computational structural genomics approach to classify ABCC6 variants using 3D models and mechanistic ontology.
Findings
Two 3D hotspots and six key functions of ABCC6 were identified that are impacted by genetic variants.
Structure-based evidence reclassified 33 variants as Likely Pathogenic and explained pathogenicity for 41% of VUS.
The approach improves diagnosis and highlights the potential of computational methods in variant classification.
Abstract
Genetic variation in ATP Binding Cassette Subfamily C Member 6 (ABCC6) can cause both pseudoxanthoma elasticum (PXE) and generalized arterial calcification of infancy (GACI). There are 930 distinct missense variants in ABCC6 reported, 87% of which are of uncertain clinical significance (VUS). New approaches are needed to mechanistically interpret and classify these VUS. We developed 3D protein models of ABCC6 in three functionally relevant conformations to calculate the structural effects of variants. We also used three-dimensional (3D) hotspot detection and developed a mechanistic ontology for critical structure-based functions of ABCC6, enabling us to categorize genomic variants. We identified two 3D hotspots and six specific functions of ABCC6 which variants impact. From this, we propose a mechanism for pathogenicity for 41% of VUS according to their impacted function. We propose…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
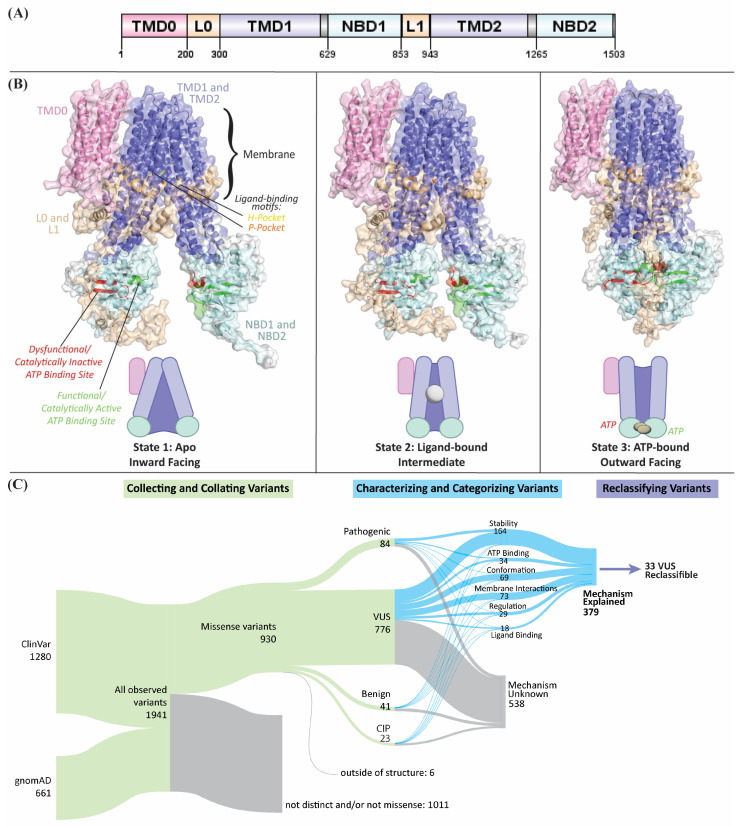 Figure 1
Figure 1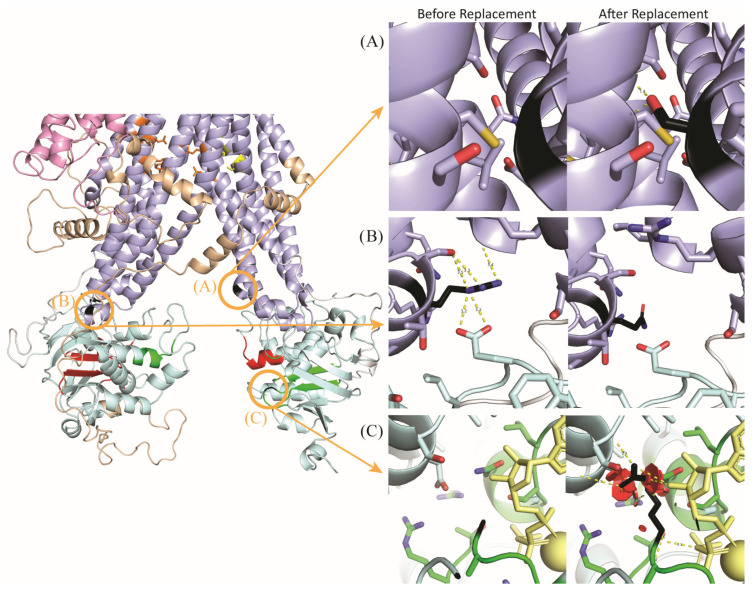 Figure 2
Figure 2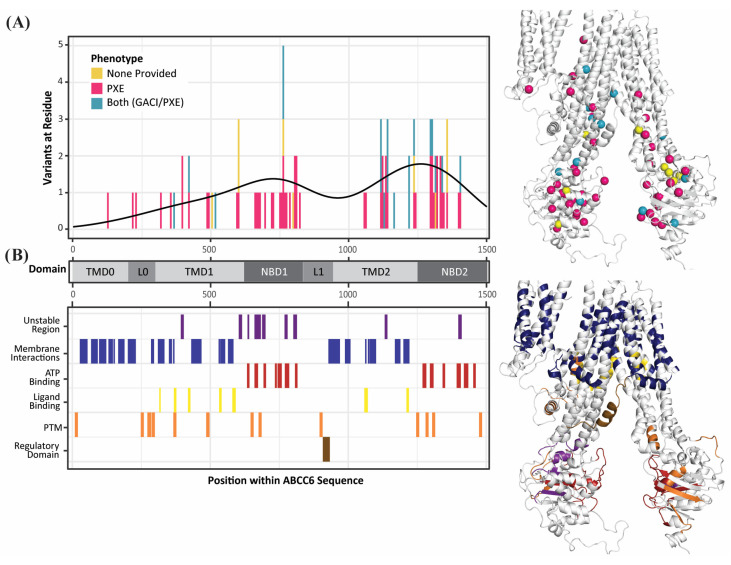 Figure 3
Figure 3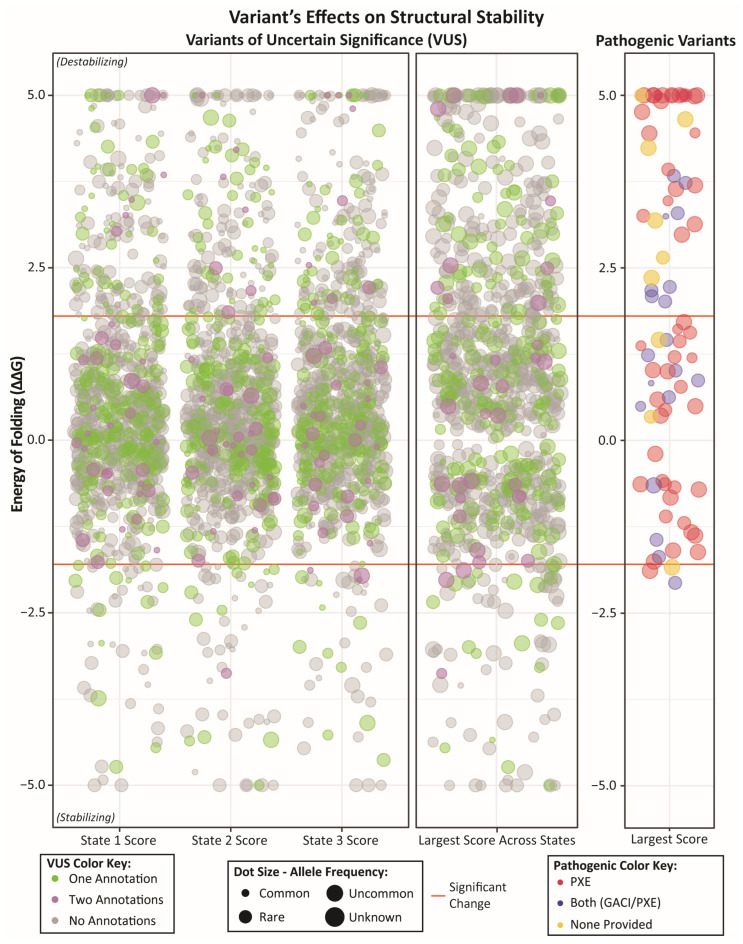 Figure 4
Figure 4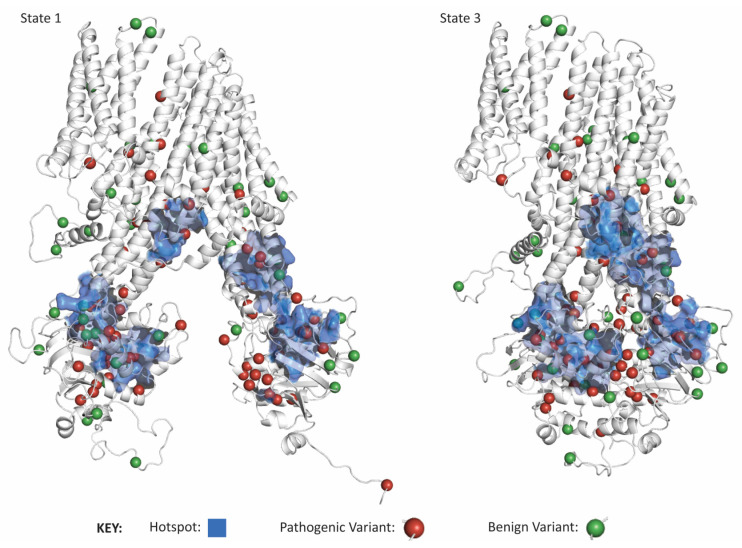 Figure 5
Figure 5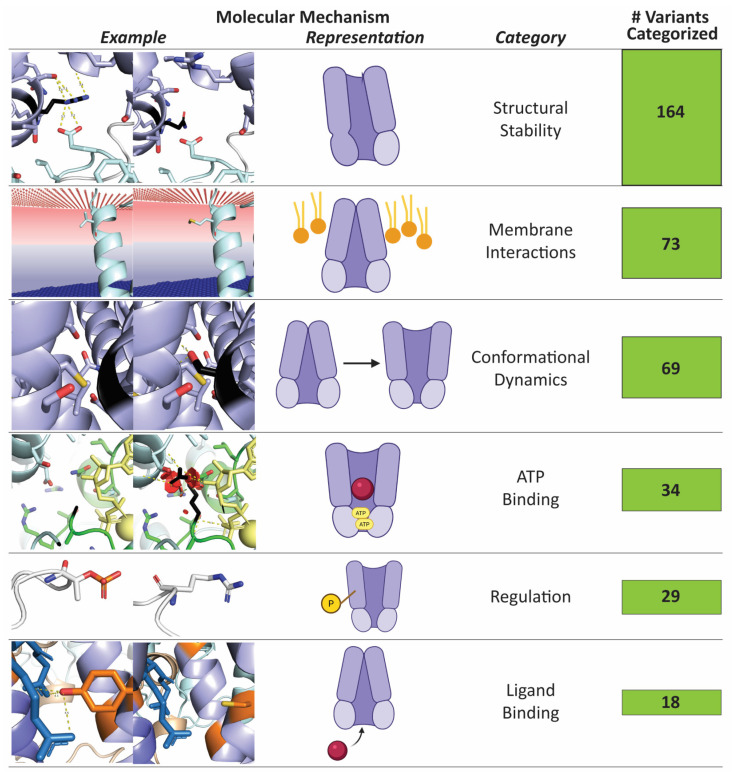 Figure 6
Figure 6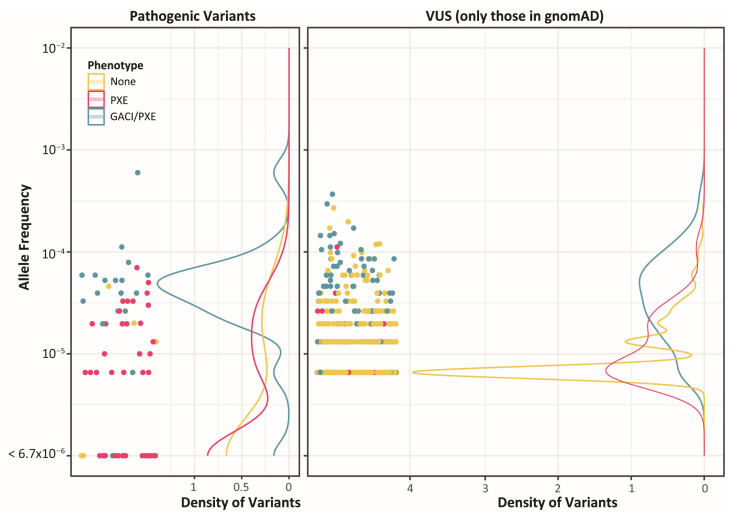 Figure 7
Figure 7- —The Linda T. and John A. Mellowes Center for Genomic Sciences and Precision Medicine at the Medical College of Wisconsin
- —Medical College of Wisconsin and the Intramural Research Program of the National Human Genome Research Institute
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDermatological and Skeletal Disorders · Connective tissue disorders research · Caveolin-1 and cellular processes
1. Introduction
ATP-Binding Cassette (ABC) proteins are among the most ubiquitous transport proteins, harnessing ATP hydrolysis to translocate substrates across vesicle and plasma membranes. While proteins in ABC subfamily C are most known for their role in drug resistance, they also cause heritable diseases when mutated [1]. Loss-of-function variants in ABCC6 (HGNC:57) are associated with the recessive disease pseudoxanthoma elasticum (PXE; OMIM #264800 and #177850) and, less frequently, Generalized Arterial Calcification of Infancy (GACI; OMIM #614473). Both diseases are characterized by ectopic calcification. In PXE, progressive calcification of the elastic fibers in the skin, eyes, and arteries cause dermal papules and angioid streaks, although PXE is typically not lethal. On the other hand, patients with GACI have extensive vascular calcification that leads to 50% mortality by six months of age [2,3]. Notably, GACI is more frequently caused by Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) deficiency. While genetic variation in ABCC6 has long been understood to underly PXE and, more recently, GACI [4,5,6,7], the molecular mechanisms affected by its dysregulation are not fully understood. This is partly because the normal physiologic function of ABCC6 remains elusive despite recent work studying the role of ABCC6 in ATP and nucleotide transport. Loss of ABCC6 has been correlated with low plasma PPi levels and decreased ATP release from cells [8,9], linking ABCC6 to ENPP1, which converts extracellular ATP into PPi, and as such, mutations in ENPP1 lead to loss of extracellular PPi [10]. However, in patients with ABCC6 variants, PPi levels do not correspond with phenotype severity [5,11], and studies suggest other factors may also be at play [12,13,14]. This lack of a precise mechanism adds to the challenge of diagnosing and treating PXE and GACI. Moreover, there are hundreds of rare variants in ABCC6 whose clinical significance is uncertain (VUS; n = 782) [15]; far fewer are clinically classified as benign (n = 41) or pathogenic (n = 84). The large number of VUS needing interpretation leaves many individuals without clear answers about their health status, limiting diagnostic and therapeutic research options.
Our work to increase the scale and rigor of computational structural genomics [16,17,18,19] and increased adoption of related approaches by other labs [20,21] demonstrate the high value of a proteogenomic approach that leverages structural bioinformatics to interpret the mechanisms of dysfunction of missense genetic variation. We anticipate that the ever expanding national and international genomics datasets will continue to increase the need for more accurate approaches to mechanistically interpreting human genetic variation.
While there is currently no full-length experimental structure of ABCC6, information from other members of its conserved family of transport proteins can be leveraged to predict the structure of ABCC6 and infer basic features of its catalytic and transport mechanisms. ABCC6 is a long-type multidrug resistance protein [22], meaning it contains two transmembrane domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2), and an initial transmembrane domain (TMD0) [23]. TMD0 is linked to TMD1 through a loop designated L0; a second loop, L1, links NBD1 to TMD2 (Figure 1A). TMDs 1 and 2 create a gated channel through the cellular membrane, open to the intracellular side at baseline, allowing ligand binding. When two ATP-Mg^+2^ bind at the interface between the two cytoplasmic NBDs, the extracellular facing conformation is formed, completing transport. ATP hydrolysis returns the protein to its initial state (Figure 1B). Thus, despite the lack of experimentally solved structures, there is considerable opportunity to apply structural bioinformatics approaches to mechanistically interpret ABCC6 genetic variation.
The current study uses a proteogenomic approach, leveraging multidisciplinary, mechanical, and functional information to characterize 924 ABCC6 missense genetic variants. Our approach includes calculating state-specific scores to harness the dynamic nature of the transport process to identify key functional regions more holistically. From these investigations, we categorize 319 (41%) VUS by their likelihood to alter specific protein features (Figure 1C). Additionally, we propose that 33 VUS can be reclassified as likely pathogenic according to their underlying molecular mechanisms. Another 134 VUS, with the additional evidence we propose, are on the cusp of a likely pathogenic interpretation. Through our process, we aim to better understand which variants may cause PXE and GACI. Finally, we anticipate that by accounting for molecular context, such as we have herein through integrative 3D modeling and biophysical characterizations, more genetic variants can be clearly categorized, functional genomics tests better designed, and clinical diagnostics improved.
2. Results
We used a proteogenomic approach [24], applying structural bioinformatics and computational biochemistry to analyze ABCC6 missense genomic variation. The full dataset contains 84 pathogenic variants from ClinVar, 776 VUS from the union of ClinVar and gnomAD, and 41 benign variants from ClinVar (Supplementary Table S1). Our study was initiated from deep structural analysis of the ten variants from our patient cohort, eight of which are pathogenic and two are VUS.
2.1. Categorizing Clinical Genetic Variants into Three Mechanisms of Dysfunction
Reviewing the variants identified in our clinical cohort revealed three main mechanisms through which genetic variants affected the protein: (1) preventing conformational changes; (2) directly destabilizing the protein structure; and (3) disrupting ATP binding (Table 1). Variants impair conformational dynamics by changing the transmembrane domain stability in one state, impacting its ability to move into the next state. For example, replacing the glycine with a serine at position 1042 creates new interactions between adjacent helices in state 3, impairing ABCC6’s ability to reset to its state 1 conformation after ATP hydrolysis (Figure 2A) (NP_001162.5:p.G1042S). On the other hand, variants that destabilize the protein structure in different regions or multiple states will not impact ABCC6’s ability to move between conformations but still damage its structural integrity and, therefore, ability to function as efficiently. For example, Arg1138 in TMD2 is necessary to stabilize the TMD-NBD joint due to arginine’s capacity to make multiple bonds with nearby residues; when arginine is replaced with glutamine (variant NP_001162.5:p.R1138Q), it loses essential bonds (such as with Glu679 in NBD1) that hold the two domains together in the proper orientation (Figure 2B). Finally, any nonconservative variant within 5 Å of the ATP binding site in 3D space will likely disrupt ATP binding by losing site-specific properties. One example is Gly1302, which is nearby (3.8 Å) to ATP. Replacing small glycine with large and charged arginine causes many steric clashes, contorting the ATP binding site and hindering ATP binding (Figure 2C) (variant NP_001162.5:p.G1302R). Overall, inspection of each variant’s effects on ABCC6’s structure and function reveals mechanisms by which all ten disease-associated variants impact ABCC6 in ways we can both measure and biochemically explain.
Guided by the above considerations and our understanding of wildtype (WT) ABCC6 functioning, we developed an algorithm for systematically categorizing the mechanistic effect of ABCC6 variants. The 10 cohort variants were run through the algorithm, and eight were predicted to have the same mechanistic effect as we had assigned through our manual study. The remaining two, NP_001162.5:p.S317R and NP_001162.5:p.T1130M, were noted above as having mild differences by our manual review but were not classified by our algorithm. This provides evidence that our new algorithm can categorize ABCC6 genetic variants with precision comparable to an in-depth manual study.
2.2. Pathogenic Variants Cluster in Three Hotspots and Mostly Affect ATP Binding or Structural Stability
We analyzed the effect of pathogenic variants on ABCC6 functionality since these are all likely to have a deleterious impact on the functioning of ABCC6. First, we analyzed whether patterns differentiate the PXE and GACI variants (Figure 3). Interestingly, TMD2 contains the largest portion of GACI/PXE variants (47%), while most variants associated with just PXE are in NBD1 or NBD2 (34% and 27%, respectively). NBD1 and NBD2 contain many ATP-binding residues, while NBD1 specifically has many canonical residues that destabilize ABCC proteins (compared to other ABC-family proteins). On the other hand, TMD2 contains many membrane-interacting and ligand-binding residues. However, one trend observed across both phenotypes was towards instability—almost half (46%) of the pathogenic variants’ most significant effect was destabilizing the protein, while less than 4% of the variants’ most considerable effect was stabilizing (Figure 4).
Next, we performed a 3D hotspot analysis of pathogenic variants to reveal any deeper pattern across variants location in 3D. Since we only had the allele count for some pathogenic variants, we counted variants in one region and the total allele count of variants in one area. Despite seeking hotspots of two different groups of variants, and in both states 1 and 3, we found hotspots in the same three regions in all analyses—one hotspot within each NBD and one spanning between the divide in the TMD bundles (Figure 5; Appendix A). These hotspots were statistically significant (p < 0.0001). Furthermore, 62 of the 82 hotspot residues (76%) were shared amongst the hotspot analyses we ran. It is worth noting that hotspots correspond to critical functions of ABCC6: the first two hotspots are in the NBDs where ATP binding and hydrolysis facilitate ligand movement out of the cell, and the third hotspot is in between the TMDs where the ligand initially binds to initiate transport, and transiently interacts with ABCC6. Thus, pathogenic genetic variations which appear heterogeneous along the linear sequence, form 3D hotspots that more homogeneously affect ABCC6’s nucleotide and ligand binding capacities.
Finally, we predicted the likely impact of ABCC6’s pathogenic variants on its core functions, categorizing over half (58%) (Supplementary Figure S1). Interestingly, more variants without a reported phenotype could be predicted than those associated with PXE or GACI. Most variants impact ABCC6’s ability to bind ATP or its structural stability—PXE variants more frequently influenced ATP binding, while variants unassociated with any phenotype more often affected structural stability. This categorization goes further than predicting variant pathogenicity by providing a mechanism by which known pathogenic variants likely contribute to PXE and GACI, while also providing a dysfunctional variant baseline for VUS comparison.
2.3. A Large Portion of Variants of Unknown Significance May Damage ABCC6 Function
We could confidently categorize a large percentage (319 variants; 41% of the total) of VUS by their likely effect on ABCC6’s core functions (Supplementary Figure S2), proposing how each likely damages ABCC6 function from a topological and biochemical perspective and, therefore, whether likely damaging effects could underly disease causality. Further, of the 168 VUS associated with either phenotype, 49% were predicted to have a specific mechanistic effect. More variants associated only with PXE were classified (65%), while fewer variants associated with both or neither phenotype were predicted (37%). Similarly, more VUS were destabilizing (27%) than stabilizing (9%) (Figure 4). Further, while state-specific average stability changes were neutral, most variants exhibited a non-zero structural stability shift in a state-specific manner. As such the main impact of many variants is on structural stability (whether global or local stability), followed by impaired membrane interactions and conformational dynamics, especially the PXE variants. Altogether, from 860 VUS, we categorize the impacted molecular mechanism for 379 variants (44%) (Figure 6).
2.4. A Lack of Clear Differences Between GACI and PXE Variants
Reviewing all 860 distinct non-benign missense variants, only one was associated with GACI alone. For the other variants, 131 were associated with PXE, and another 111 were associated with both GACI and PXE diagnoses (which we will refer to as GACI/PXE variants). Further, for both the pathogenic variants and VUS, the allele frequency was, on average, higher in variants associated with GACI/PXE than those associated with just PXE or neither phenotype (Figure 7). Despite apparent differences in the clinical severity and allele frequency between the variants responsible for PXE and those responsible for GACI, the lack of genetic variants that are unique to GACI makes it challenging to delineate PXE-causing variants from GACI-causing variants.
2.5. Structural Bioinformatics Links VUS Mechanisms to Pathogenicity
We find 33 VUS where structural calculations provide a robust level of evidence that could be used to interpret patient variants, concordant with ACMG guidelines. Specifically, in this manuscript we provide novel, mechanistic-based evidence for PM1 (i.e., “Mutational hot spot or well-studied functional domain without benign variation”) based on our 3D hotspots and functional annotations. Further, we propose new PP3 (i.e., “multiple lines of computational evidence support a deleterious effect on the gene/gene product”) evidence that is distinct from in silico tools [25,26] and based on the specific mechanism affected (Supplementary Table S1; Appendix B). Using this novel evidence along with readily and publicly available annotations of PM2 (i.e., “absent in population databases”), PM5 (i.e., “novel missense change at an amino acid residue where a different pathogenic missense change has been seen before”), and PP4 (i.e., “patient’s phenotype or FH highly specific for gene”), we make a simple and illustrative comparison wherein the number of likely pathogenic variants could increase by 39% (Table 2). Furthermore, another 141 VUS will only require one additional line of evidence to be reclassified as likely pathogenic, which would likely be readily available for a familial case. Similarly, one variant with conflicting interpretations of pathogenicity would now have enough evidence to potentially be reclassified as likely pathogenic, and 11 would require only one additional line of evidence. Furthermore, using the same lines of evidence as with the VUS, our data would be consistent with classifying 51 of the 84 pathogenic or likely pathogenic variants in ClinVar as likely pathogenic, and the remaining 33 variants only need one more line of evidence to reach this status. This strong congruency, before the necessary addition of patient-specific clinical data, demonstrates the strong likelihood for computational structural genomics analyses to enhance the classification and interpretation of human genetic variation.
We propose that our analysis of variants’ effects on key functions of ABCC6 is a robust and biophysically justified evidence of alteration, which could be a novel way of reframing the PM1 criteria as altering a “key function” in three dimensions—energetically, quantitatively, and at atomic resolution—rather than a “key functional site” as defined by sequence data and low throughput mutational experiments. If our categorization were used as PM1, and a combination of CADD and REVEL used as PP3, 100% of benign variants and 94% of pathogenic variants would be classified the same as ClinVar (achieving 96% accuracy), and 80 VUS would have enough evidence to be reclassifiable as Likely Pathogenic (Supplementary Table S2), again demonstrating the potential power of this approach.
In summary, this study elucidates complex mechanisms underlying ABCC6 dysfunctions and underscores the need for increasing our understanding of population-specific genetic variations that are applicable to n = 1 cases. We conclude that integrating biophysical and biochemical metrics, proteogenomic approaches, and computational algorithms has substantially enhanced our comprehension of ABCC6 variants. This understanding builds a trajectory for more accurate and mechanistic clinical diagnoses, leaving less ambiguity for patients.
3. Discussion
Currently, less than 14% of ABCC6 alleles reported in ClinVar and gnomAD have a confident clinical interpretation. Among variants with an interpretation, there is no clear underlying disease mechanism. These gaps in knowledge make variant interpretation and treatment difficult. To further investigate this issue, we used a multi-tiered proteogenomic approach to study ABCC6 variants. We analyzed the variants in three steps to characterize their molecular context and mechanistic effect. Our findings show that our approach is effective and insightful in determining the functional mechanism of ABCC6 genetic variants. By encoding this process as an algorithm, we mechanistically categorized 41% of VUS to high confidence (See Supplementary Table S1). Further, with the information gathered, we also explored differences between variants associated with GACI and PXE—differences that may help us better understand the relationship between the two fundamentally similar yet distinct disorders. Projecting currently identified pathogenic variants onto our 3D structures enabled us to find mutational hotspots. Finally, we provide evidence for five ACMG categories across all 924 variants based on novel structural biology insights, which can be used to aid in classification. Our study adds to what is currently known about both the mechanistic effect of ABCC6 variants and the deleteriousness of VUS, which can help clinicians better interpret patient genotypes.
Previous studies of ABCC6 variants have relied on a singular homology model [27,28], characterized up to tens of variants [29,30,31,32,33,34,35], or have relied on purely clinical and/or functional information [36], revealing how ABCC6 mutations impair membrane localization, regulation, NBD structural stability, and ligand transportation. Building off those, this is the first study of ABCC6 to describe nearly a thousand variants using multi-state models and extensive mechanistic data, expanding the categories of dysfunction to include general stability and conformational dynamics. This integrative approach enables us to characterize far more variants than we could if we used one model or only functional information. Furthermore, by developing an algorithm to perform our analysis and evaluate evidence of ACMG classifications, and by providing the code to do so (Supplemental Text), any additional variants can be analyzed in the same fashion, making this study both repeatable and applicable to any possible variant in ABCC6 (besides those in the six initial residues not included in our modeled structures). Our results demonstrate the utility of our multi-tiered proteogenomic approach and lead to novel conclusions about ABCC6.
Few studies have directly compared PXE-causing variants to GACI-causing variants in ABCC6; therefore, our study boasts new information about ABCC6 variants’ disease associations. We discovered few variants observed exclusively in GACI patients and few differences in mechanisms between variants associated with PXE and those associated with GACI/PXE. This may, in part, be due to lack of specificity in ClinVar variant reporting, especially when a variant is added by large-scale genetics companies which don’t provide phenotypic information. Further, we found that GACI/PXE variants have a higher allele frequency. Since GACI is less common than PXE [37,38], GACI would be more statistically likely to be associated with more common variants if all variants were equally likely to lead to GACI on their own. Altogether, this suggests that the molecular differences between PXE and GACI are not caused by specific variants, which is supported by the divergent diagnoses, sometimes even within siblings [39]; instead, other distinguishing factors are likely at play, such as a genetic or epigenetic modifier, a metabolic environment, a change in regulation, an external stressor, etc.—as current studies are investigating [40]. This directs future studies of ABCC6 to focus on patient-to-patient differences, rather than variant-to-variant differences, in differentiating GACI and PXE.
This study also represents a significant advance in understanding genetic variants in ABCC6 through methodologies that could help with other rare disease genes. Our approach predicted the mechanistic effect of 319 VUS that likely impair ABCC6 function enough to cause protein dysfunction. These six functions cover an array of variant effects, which is critical given recent research revealing that, while structural stability is important, many other detriments can cause disease [41]. While we maintain a similar precision (96%) to current best-in-class pathogenicity prediction algorithms, such as CADD and AlphaMissense (100%), and Sherloc (100% in prior study [36]), our accuracy (72%) is only slightly lower than CADD’s (86%) but higher than AlphaMissense (67%). This is due to using cautious thresholds to prevent false positives, as well as the lack of patient-specific information. However, the mechanistic nature of our structure-based classification provides additional value which could also inform potential treatment options. It also enables hypothesis testing, an improvement over standard genomic in silico scores—in fact, 30 of the variants we studied have already been tested in vitro [29,31,33,42]. Our predicted functional impact was supported in 17 of the 25 comparable variants (Supplementary Table S3), with our methodology not identifying a mechanism on six variants which were dysfunctional, possibly due to overly cautious thresholds or lack of information. Further functional study designs can be guided by the results of this study.
Another significant contribution of this study is its methodology, which can be applied to many other proteins and variants. Indeed, our study takes a new approach to genomic variant analysis and interpretation, focusing on the mechanistic effect of a variant on the translated gene product rather than the likelihood of pathogenicity. The damaging potential of a variant is clearly related to its probability of pathogenicity. However, our current calculations do not capture all contexts (such as changes to protein folding or impacts on any potential protein interactions that have yet to be discovered). Creating an algorithm specifically for ABCC6 that considers a large amount of specific contextual information enables even the rare and singleton variants to be understood—something statistics-based approaches struggle with. Furthermore, our study revealed clusters of pathogenic variants within three main hotspots, which are spatially near functionally significant zones of ATP binding and ligand binding. This demonstrates the close relationship between a variant’s pathogenicity and the protein’s function—making understanding protein function especially critical in advancing better variant interpretation. These hotspots are similar to those identified by prior studies, [32,33,43], which identified hotspots in one nucleotide binding domain, as well as at the interfaces between TMDs and NBDs and between TMDs (such as the ligand binding pocket), providing further granularity to this ongoing effort. Our hotspot analysis and functional annotations enabled much of our variant reclassification and will continue to provide PM1 evidence for further reclassification of VUS. Overall, our results demonstrate that a protein-specific structural bioinformatics approach like ours helps us better understand the mechanisms of pathogenicity for VUS, and this methodology can be used to study variants of any protein with some known structural and functional information.
Through future studies, additional cell-based data will provide further understanding of the differences between PXE and GACI. Using multiple functional conformations to structurally characterize variants, integrated with additional functional genomics information, can create more specific and accurate studies. Results from the current work can guide future studies of other variants with the methods described herein, leading to better interpretation of patient genomes and, eventually, more successful treatment of both diseases.
4. Materials and Methods
4.1. Integrative Three-State Protein Structure Modeling Captures Details of ABCC6 Function
There are no full-length experimentally determined models of ABCC6. We used integrative modeling to generate the first models of ABCC6 for use in scoring genomic variation. ABCC6’s closest homologous protein, ABCC1, has a series of 3 bovine models published in the Protein Data Bank (PDB) [44,45,46] that show the range of motion ABCC1 goes through at the un-liganded inward-facing conformation to an intermediate conformation with substrate bound, and then to the outward-facing conformation with ATP bound at the hydrolytic domain. After these three steps, ABCC1 hydrolyzes ATP to return to its apo conformation. Because each structure highlights an essential step in ABCC1’s function, and ABCC6 is thought to have a similar series and mechanism of motion, we used each structure as a template in Modeller [47] to generate 3D models of ABCC6 in 3 functional states. However, the ABCC1 models (and consequently ABCC6 models) lacked the TMD0 domain of ABCC6. YCF1, the yeast ortholog of ABCC1, has two structures on the PDB [27,48], which include a well-resolved TMD0 and has better resolution on L0 and L1 than ABCC1’s structures. Therefore, these two structures were used as templates with Phyre [49] to create two models of ABCC6 in the inward-facing conformation (state 1) and intermediary conformation (state 2). There is no structure of outward-facing (state 3) YCF1, but TMD0 and L0 had negligible conformational differences between states 1 and 2, so an improved series of models were created by replacing the TMD0 and L0 domains from the ABCC1-template structures with the YCF1-template state one domains, and replacing L1 in states 1 and 2 with the YCF1-template model L1 (but retaining the ABCC1-template L1 in state 3) (Figure 1B). PyMOL v2.5 was used to visualize structures [50]. These steps combined generated molecular models for mechanistic interpretation of ABCC6 genetic variations.
4.2. Genomic Variation Collection and Annotation
We generated three datasets of genomic variation. First, our team selected ten missense variants from our clinical cohort with deep clinical phenotyping data. Second, we assembled 1280 ABCC6 variants from the May 2023 ClinVar release [15] and 661 missense ABCC6 variants from gnomAD v3.1.222 using the canonical transcript (ENST00000205557). Nomenclature was validated with VariantValidator [51]. Annotations about each variant, such as associated phenotype, pathogenicity, and allele frequency, were provided by ClinVar and gnomAD, respectively (when available). Ensembl’s [52] Variant Recorder was used to obtain variant HGVS Protein labels for ClinVar variants. These data were collated into one dataset of 930 distinct missense variants to compare information and context on each variant. We divided this dataset into two subsets: (1) all 84 pathogenic (and likely pathogenic) missense variants from ClinVar as true positives and (2) the 805 missense variants that are annotated as variants of uncertain significance (VUS) or variants with conflicting interpretations of pathogenicity (CIP), or those for which there were no clinical annotations available. However, six variants (P4H, A9E, G12E, E18D, P21S, and A23T) were at amino acids not shown in our structures (which began at residue 26). These variants were excluded from analysis, leaving 84 pathogenic missense variants, 776 VUS, 41 benign (and likely benign) variants, and 23 variants with CIP to be analyzed.
4.3. Annotating Motifs, Biochemical Regions, and Post Translational Modification Sites
We first gathered sequence-based information about ABCC6’s motifs and post-translational modifications (PTM) to understand the mechanistic effects of the above variants. First, within the nucleotide-binding domain, there are five highly conserved motifs across the ABC family: Walker A (also called P-loop or phosphate-binding loop), Walker B, Switch region (also called H-loop or histidine loop), signature motif (also called C-loop), and Q-loop. These motifs, among other residues, form the nucleotide-binding pocket, which classically consists of the Walker A and B, Q-loop, and H-loop from one NBD and the signature motif of the other NBD [53,54,55]. Variants within these motifs, or spatially supporting each motif, could affect motif functions. This has been documented already, as ABC Subfamily C (ABCC) proteins all carry a mutation of Glu778 to Asp in the Walker B sequence of NBD1, causing decreased efficiency of ATP hydrolysis for that ATP binding site [44,56,57]; hence, NBD1 is considered dysfunctional. Similarly, ABCC proteins are missing 10–13 highly conserved residues in NBD1, residues which in other ABC proteins stabilize the TMD-NBD interface; due to this loss of essential contacts, ABCC proteins are less stable in this region [45,57]. As such, variants in these regions are likely to impact the already-unstable NBD1-TMD interface or the functionally-critical Walker motifs.
Linker regions are also regulatory domains. Within L1, also sometimes called the regulatory domain or R-domain, lies a highly conserved phosphorylation site at S902, where studies in YCF1 (a yeast homolog for ABCC1) demonstrated PKA phosphorylation of that site to regulate YCF1 activity by inducing structural changes upon phosphorylation. L1 also contacts NBD1 through Q906 binding with Q715, stabilizing and regulating activity [48]. Beyond the PTMs in L1, residues 911–931 are also known to be of regulatory importance [58]. Within L0, a CK2 phosphorylation site is highly conserved at S244, which negatively regulates substrate transport, and a predicted basolateral localization sequence aids in proper localization [48,59]. All these residues, as well as residues within three amino acids of a PTM, were considered of regulatory importance. Finally, there are 13 other PTMs without an experimentally-discovered function suggested by both iPMTnet [60] and PhosphoSitePlus [61], 9 of which are phosphorylations, two acetylations, one ubiquitination (on the same amino acid as one of the acetylations), and one n-glycosylation. Any variants sequentially or spatially near these PTMs were noted as well.
4.4. Structure-Based Biophysical Annotations and Fold Stability
Since the ABCC1 substrate and ATP binding pockets are well-defined [44,45,56,57,62], sequence alignment was used to predict the ABCC6 substrate binding sites. To obtain further 3D contacts for both ATP and the ligand, we aligned the template ABCC1 structures with our ABCC6 structure in states 2 and 3 to confirm the homology-based prediction of ligand-binding pockets. We defined the pockets as the union of conserved ligand-binding sites, ATP-binding sequence motifs, and all residues within 5 Å of aligned ABCC1 experimentally-determined ligand positions.
The membrane-spanning portion of ABCC6 was predicted with the PPM 3.0 webserver [63] and all residues within this region and an accessible surface area of >50% were tagged as lipid-interacting. We also noted three positively charged helices, which rest upon the membrane’s outer layer at residues 204–208, 217–228, and 931–943. These residues were tagged for their likely role in charge-stabilizing membrane head group interactions.
FoldX [64] was used to obtain the change in folding free energy (ΔΔG_fold_) for each variant compared to the wildtype structure. The same procedure was performed with all three conformations as input and across all pathogenic variants from all three cohorts. For the 10 cohort variants, FrustratometeR [65] was also used to obtain the change in folding frustration energy. Finally, for all variants, the PAM30 score for the replacement was obtained, as well as the difference in sidechain hydrophobicity using the Peptides R package [66] and the Kyte–Doolittle scale [67].
4.5. Discovery of 3D Genetic Variant Hotspots
We determined amino acid contact maps using those within 10 Å of each amino acid in the state 1 and state 3 structures. Since 39 of the 84 pathogenic variants are also reported in gnomAD, we performed two rounds of hotspot analysis. First, we weighted variants by their population incidence by summing the allele counts for all gnomAD-reported variants within 10 Å of each residue. Second, we weighted distinct variants by summing the total number of variants (out of the 84 total pathogenic variants) within 10 Å of each residue. The residues with 15 or more alleles and 7 or more variants, respectively, were considered within the hotspot since all residues with that number of alleles/variants had a p-value < 0.0001. To calculate statistical significance, we first made two null sets with the same number of randomly selected variants as each of our observations (total allele count of 269 and 84 total variants, respectively) and repeated a similar procedure, permuting selection 10,000 times to obtain the number of times that randomly distributed variants were more densely populated around each residue than our observations. Dividing that number by 10,000 gave an empirical p-value.
4.6. Categorizing Mechanistic Effect of Variants
To characterize the range of effects that genetic variants have on ABCC6, we describe six essential aspects of ABCC6 function: ATP Binding, Ligand Binding, Membrane Interactions, Regulation, Conformational Dynamics, and Functional Stability (Table 3). These six categories of functional effect were chosen based on our study of wild-type ABCC6 and a cursory review of the variants’ impacts. They formed the basis of our ABCC6-specific context-guided variant effect algorithm—a unique approach to variant effect interpretation that focuses on understanding the mechanism of variant impact rather than correlating features to clinical information. This decision-tree algorithm uses all the annotations and scores described in prior sections to categorize variants into one of the six categories (see Supplementary Materials for the code). Its rules are logically derived: a series of metrics were chosen to define each function, and thresholds set for those metrics (Suppmentary Text S2). Then, each function is tested based on order of specificity—for example, if a variant is destabilizing, and destabilizing to the ATP binding region, it is impacting ATP binding specifically. We tested our algorithm on the cohort variants and compared it with the manual categorization we performed, then applied it to missense pathogenic variants, benign variants, and VUS. R packages ComplexHeatmap [68] and Tidyverse [69] were used in our analysis. Finally, other pathogenicity prediction scores from CADD [26], REVEL [25], PolyPhen2 [70], AlphaMissense [71], and SIFT [72] were obtained through their respective servers or databases. The threshold for which variants were predicted to be pathogenic was set at their respective recommended thresholds: ≥20 for CADD (using the PHRED score) [26], ≥0.5 for REVEL [25] and PolyPhen2 [73], ≥0.564 for AlphaMissense [71], and ≤0.05 for SIFT [73].
4.7. Reclassifying Variants of Uncertain Significance
The American College of Medical Genetics and Genomics (ACMG) guidelines for interpreting variant effects were used to organize evidence for VUS classification [74]. We propose that our computational structural genomics approach can provide evidence types PM1 and PP3. How to apply computational structural genomics to these additional lines of evidence for clinical interpretation is a crucial advancement from the current study. We directly compare our categorization of variants to the existing classification (Figure 1C and Table 3). For additional comparisons to our novel variant classification approaches, we also used evidence lines PM2, PM5, and PP4, established in the field and obtainable through publicly available data. For PM1, variants were noted that reside within the well-defined Walker A motif, Walker B motif, Signature Loop, H-loop, and Q-loop [53,54,56] as well as variants residing within the variant-rich three 3D-hotspots identified herein. For PP3, all variants predicted by our methodology to impact key ABCC6 functionalities were noted. For PM2, variants that were present within the ClinVar database but not gnomAD were noted [15,75], as well as those with an allele frequency less than 0.33% and at residues past 250. The allele frequency was chosen as 0.33% since the carrier frequency for PXE is expected to be between 1 in 150 and 1 in 300 people [76], and the carrier frequency for GACI is expected to be around 1 in 200 people [77]. Residues before 250 were not included since the coverage of residues 0–250 in gnomAD was lacking, i.e., the fraction of individuals with coverage over 30 was less than 50%. The coverage of ABCC6 within gnomAD v3 was confirmed to be of sufficient read depth. For PM5, all variants at the same residue position as a pathogenic variant from the ClinVar database were noted. Finally, for PP4, all variants with an associated phenotype of PXE, GACI, or both in ClinVar were noted. The 2015 ACMG guidelines [74] were used to count evidence and determine whether sufficient evidence is present for potential reclassification.
5. Conclusions
With a large range of phenotypes emerging from variants in ABCC6, determining the deleteriousness of genetic variants and their mechanisms for impairing function is critical for diagnostic and therapeutic research. The current study employs a novel multi-tiered approach to ABCC6 variant analysis and identifies the underlying molecular effects for 60% of pathogenic variants and 43% of VUS, identifying additional information that supports 33 VUS to be reclassified as Likely Pathogenic. In a sense, this is the first mechanistic proteogenomic ontology for ABCC6. Approaches such as we report here not only enable more genetic diagnoses, but also provide a new kind of information for clinical interpretation that is applicable to the one-of-a-kind genetic variation that frequently underlies rare diseases. The most common function affected was structural stability, yet the other five categories cumulatively account for 57% of variant classification, highlighting the importance of more holistic consideration of the structural implications of genetic variants on their encoded products. This study supports a different way of thinking about variant interpretation in general: the mechanistic effects of DNA changes can be enhanced by closely considering the intersection of structure and function.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1StefkováJ. Poledne R. Hubácek J.A. ATP-binding cassette (ABC) transporters in human metabolism and diseases Physiol. Res.20045323524310.33549/physiolres.93043215209530 · doi ↗ · pubmed ↗
- 2O’Neill M.J.F. Online Mendelian Inheritance in Man, OMIM. Johns Hopkins University, Baltimore, MD. MIM Number: 610508. OMIM 2010 Available online: http://omim.org/entry/610508(accessed on 16 May 2022)
- 3Stelzer G. Rosen N. Plaschkes I. Zimmerman S. Twik M. Fishilevich S. Iny Stein T. Nudel R. Lieder I. Mazor Y. The Gene Cards Suite: From Gene Data Mining to Disease Genome Sequence Analyses Curr. Protoc. Bioinform.2016541.30.11.30.3310.1002/cpbi.527322403 · doi ↗ · pubmed ↗
- 4Bergen A.A. Plomp A.S. Schuurman E.J. Terry S. Breuning M. Dauwerse H. Swart J. Kool M. van Soest S. Baas F. Mutations in ABCC 6 cause pseudoxanthoma elasticum Nat. Genet.20002522823110.1038/7610910835643 · doi ↗ · pubmed ↗
- 5Nitschke Y. Baujat G. Botschen U. Wittkampf T. du Moulin M. Stella J. Le Merrer M. Guest G. Lambot K. Tazarourte-Pinturier M.F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP 1 or ABCC 6Am. J. Hum. Genet.201290253910.1016/j.ajhg.2011.11.02022209248 PMC 3257960 · doi ↗ · pubmed ↗
- 6Ferreira C.R. Kintzinger K. Hackbarth M.E. Botschen U. Nitschke Y. Mughal M.Z. Baujat G. Schnabel D. Yuen E. Gahl W.A. Ectopic Calcification and Hypophosphatemic Rickets: Natural History of ENPP 1 and ABCC 6 Deficiencies J. Bone Miner. Res.2021362193220210.1002/jbmr.441834355424 PMC 8595532 · doi ↗ · pubmed ↗
- 7Li Q. Brodsky J.L. Conlin L.K. Pawel B. Glatz A.C. Gafni R.I. Schurgers L. Uitto J. Hakonarson H. Deardorff M.A. Mutations in the ABCC 6 gene as a cause of generalized arterial calcification of infancy: Genotypic overlap with pseudoxanthoma elasticum J. Investig. Dermatol.201413465866510.1038/jid.2013.37024008425 PMC 3945730 · doi ↗ · pubmed ↗
- 8Jansen R.S. Küçükosmanoglu A. de Haas M. Sapthu S. Otero J.A. Hegman I.E. Bergen A.A. Gorgels T.G. Borst P. van de Wetering K. ABCC 6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release Proc. Natl. Acad. Sci. USA 2013110202062021110.1073/pnas.131958211024277820 PMC 3864344 · doi ↗ · pubmed ↗