Molecular Basis of Adenomatous Gastrointestinal Polyposis Syndromes: Role of Pathogenic and Benign Variants in Disease Onset
Francesca Cammarota, Valeria D’Agostino, Chiara Capasso, Francesca Duraturo, Valentina D’Angelo, Giovanni Battista Rossi, Paola Izzo, Rosario Vicidomini, Mimmo Turano, Marina De Rosa

TL;DR
This study explores how genetic variants contribute to gastrointestinal polyposis syndromes and their role in disease onset and progression.
Contribution
The study identifies benign genetic variants that may partially contribute to disease onset or act as phenotypic modifiers.
Findings
Germline pathogenic variants were found in 55% of affected patients.
MUT+ patients had earlier disease onset and more polyps than others.
Benign variants in APC and POLD1 were linked to altered gene expression.
Abstract
Background: Colorectal cancer (CRC) is the third most diagnosed type of cancer and the second leading cause of cancer-related death. However, the increase in CRC incidence observed over the last 50 years has been accompanied by an overall reduction in mortality thanks to improved diagnostic strategies, patient follow-up, and more targeted therapies. Gastrointestinal adenomatous polyposis syndromes are a group of hereditary syndromes that predispose individuals to gastrointestinal tumors. These syndromes, characterized by the onset of gastrointestinal adenomas, are genetically heterogeneous. Methods: We analyzed 60 subjects with clinical suspicion or diagnosis of polyposis using next-generation sequencing (NGS). An additional 20 healthy individuals, all negative for pathogenic variants, were included in the study as a control population. We also performed bioinformatic analyses to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
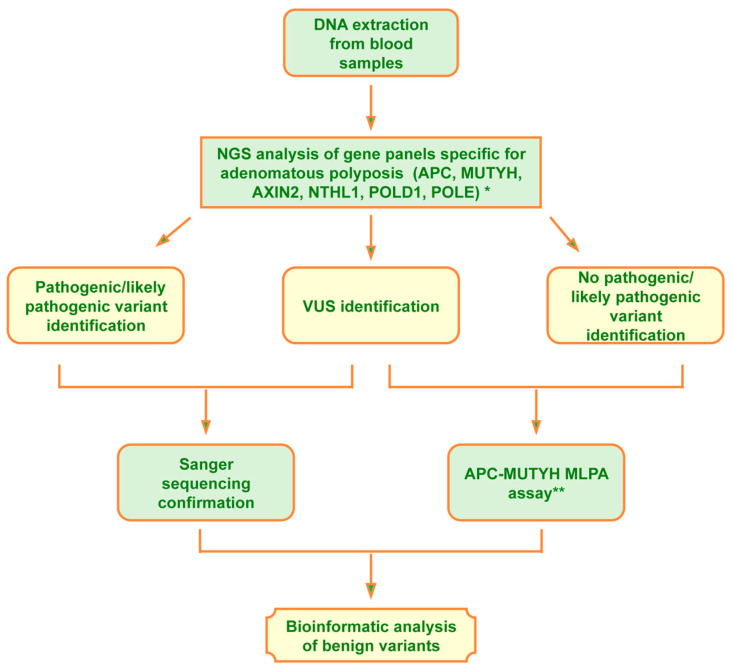 Figure 1
Figure 1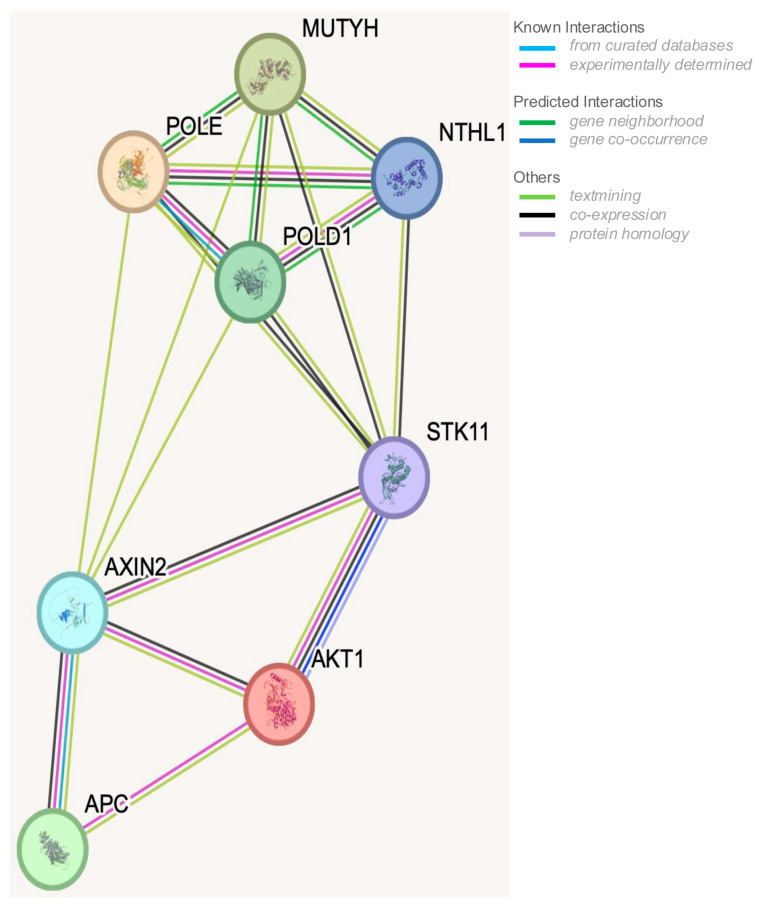 Figure 2
Figure 2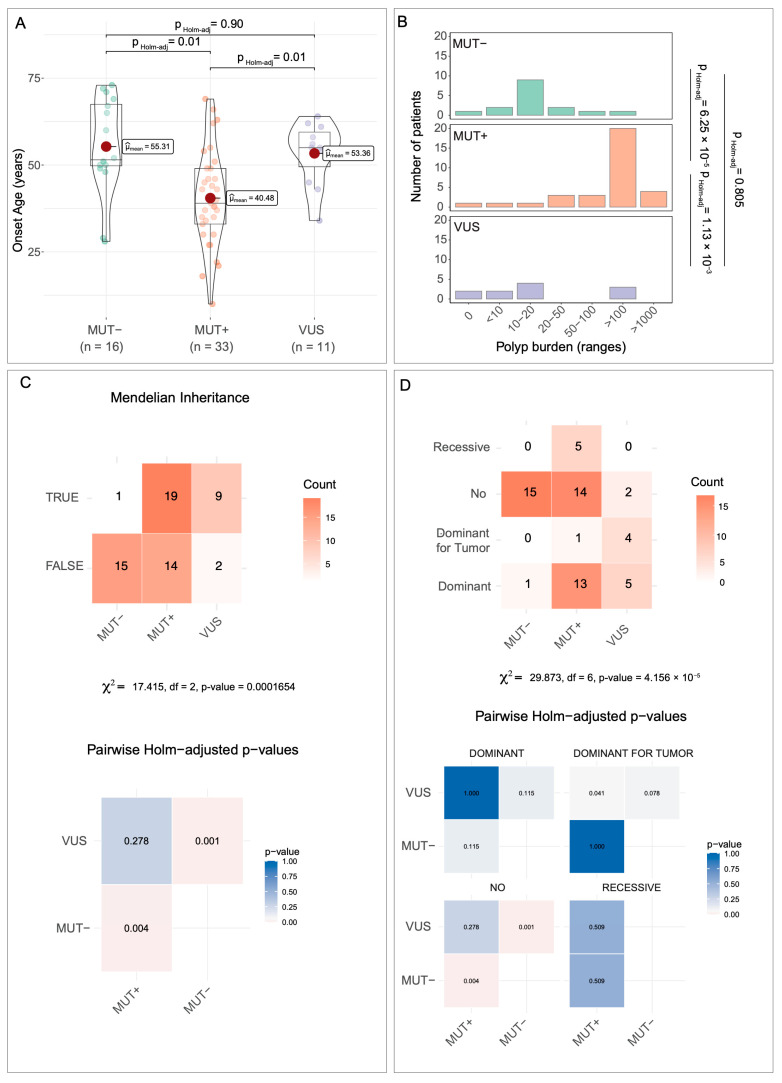 Figure 3
Figure 3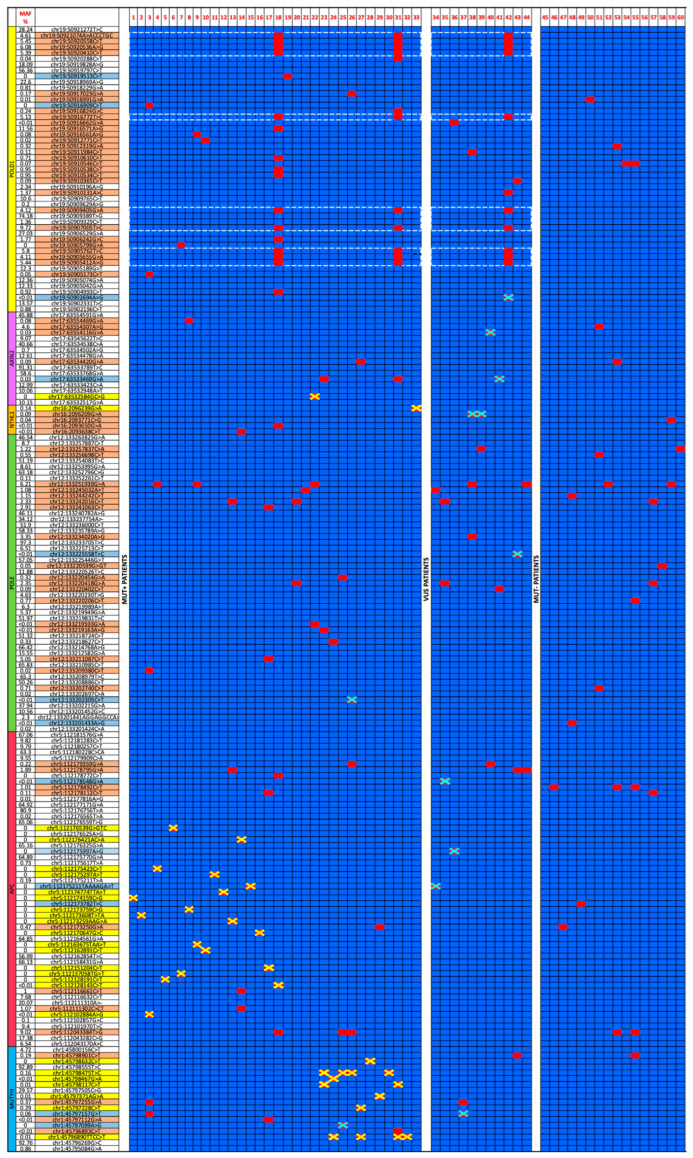 Figure 4
Figure 4- —Università degli Studi di Napoli “Federico II,”
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic factors in colorectal cancer · Colorectal Cancer Screening and Detection · Gastric Cancer Management and Outcomes
1. Introduction
Familial colorectal polyposis syndromes are a group of very rare hereditary disorders that are both phenotypically and genotypically heterogeneous and predispose individuals to colorectal tumor development [1,2,3,4]. Based on the histological characteristics of the polyps, colorectal polyposis syndromes are classified into adenomatous and hamartomatous types.
Adenomatous polyposis syndromes include familial adenomatous polyposis (FAP), attenuated FAP (AFAP), MUTYH-associated polyposis (MAP), and polymerase proofreading-associated polyposis (PPAP) [5].
Hamartomatous polyposis syndromes mainly include Peutz–Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS) [6,7], and PTEN hamartoma tumor syndrome (PHTS) [6,8,9,10].
MAP is inherited in an autosomal recessive manner, whereas all other syndromes are reported to follow an autosomal dominant inheritance pattern [11,12].
Although hereditary polyposis syndromes are classically described as monogenic disorders, nearly fully penetrant and genotype–phenotype correlations are reported, and inter- and intra-familial phenotypic variability is also described. This heterogeneity may result from modifier alleles, somatic mutations, mosaicism, or other genetic and environmental factors and may complicate both diagnosis and clinical management [5].
The genes most commonly associated with adenomatous polyposis syndromes include APC, MUTYH, NTHL1, POLE, POLD1, and AXIN2 [2,13,14]. Conversely, the genes mainly implicated in the onset of hamartomatous polyposis syndromes are STK11, PTEN, BMPR1A, SDHB, SDHD, SMAD4, AKT1, ENG, and PIK3CA [2,3,15,16,17].
FAP exhibits a broad phenotypic spectrum. The classic form is characterized by the development of hundreds to thousands of polyps, typically beginning around the age of 20. If untreated, these polyps inevitably progress to colorectal cancer, necessitating prophylactic colectomy. The attenuated form of FAP (AFAP) is marked by a later onset (around age 40) and a reduced polyp burden, usually fewer than 100 polyps. FAP patients often develop neoplasms in the upper gastrointestinal tract, such as gastric, fundic, duodenal, and ampullary adenomas, which represent the second leading cause of death after colorectal cancer (CRC). Other extraintestinal manifestations of FAP include osteomas, tooth anomalies, congenital hypertrophy of the retinal pigment epithelium (CHRPE), desmoid tumors, and extraintestinal tumors, such as thyroid, liver, bile duct, and central nervous system cancers [18,19,20].
MAP is characterized by the development of approximately 10–500 colorectal adenomas, with a lifetime risk of CRC between 43 and 48 years; the risk approaches 100% beyond the age of 48 [21]. On the other hand, monoallelic variants of MUTYH are associated with a moderate increase in CRC risk (1.5–2-fold), especially among individuals with a first-degree relative affected by CRC [21]. Individuals with MAP also face an elevated risk of duodenal cancer and non-melanoma skin cancer, as well as ovarian, bladder, and possibly endometrial cancers [22].
Individuals with PPAP may present with autosomal dominant inheritance, classical, or attenuated polyposis, CRC, and other somatic hypermutation-related tumors, even in the presence of a functioning DNA mismatch repair (MMR) system [14], including colorectal, endometrial, ovarian, breast, brain, and upper GI tumors.
The spectrum of adenomatous polyposis syndromes has recently been expanded to include two rare autosomal recessive conditions caused by biallelic mutations in NTHL1, a DNA glycosylase involved in base excision repair, and in MSH3, a gene involved in the MMR pathway [14,23]. Individuals carrying biallelic NTHL1 pathogenic variants frequently develop multiple independent tumors, highlighting the need for intensive, lifelong, and multi-system surveillance [22].
CRC screening has led to marked reductions in both cancer incidence and mortality over the past two decades [24]. In addition, family history, tumor histology, and molecular characterization are crucial for identifying individuals predisposed to CRC and to implement appropriate surveillance and treatments. Since differential clinical diagnosis can be difficult because overlapping features exist between polyposis syndromes, molecular diagnosis is pivotal for accurate classification and appropriate clinical management [20,25,26].
The primary aim of this study was to elucidate the molecular basis underlying the onset of familial adenomatous gastrointestinal polyposis syndromes and to identify the genes and mechanisms involved in their pathogenesis.
Achieving these goals will not only improve diagnostic accuracy but facilitate the identification of novel therapeutic targets for more effective diseases management.
We also sought to explore genotype–phenotype correlations by defining the clinical features of patients carrying or not carrying pathogenic variants or VUS (variants of uncertain significance), evaluating age at onset, number of colorectal polyps, and inheritance pattern (presence/absence of Mendelian autosomal inheritance). To this end, statistical analyses were performed on the studied population cohort, which was divided into the following three groups:
- Patients carrying a pathogenic/likely pathogenic germline variants (MUT+);
- Patients carrying a VUS germline variant (VUS);
- Patients without any germline variant, neither pathogenic nor VUS (MUT-).
As a secondary aim, we explored the potential contribution of benign variants, hypothesizing that such variants could lead to gene alteration through an additive effect, thereby contributing to disease onset. Previous studies, particularly genome-wide association studies, have shown that common low-penetrance variants, often classified as benign, can cumulatively modulate CRC risk [27].
2. Materials and Methods
2.1. Patients and Samples
Sixty subjects with clinical suspicion/diagnosis of adenomatous polyposis or subjects carrying pathogenic variants in genes associated with adenomatous polyposis syndromes without clinical evidence of polyposis but showing personal and familial cancer aggregation were enrolled in this study. Individuals heterozygous for monoallelic pathogenic variants in genes associated with hereditary recessive polyposis were included to explore incomplete or low-penetrance phenotypes and to reduce selection bias toward classical polyposis presentations. All probands included in this study were referred for molecular screening after a careful investigation of clinical history by a specialized clinician and genetic counseling. Inclusion criteria were the presence of multiple gastrointestinal adenomas and/or other neoplasm associated with the disease and/or positive family history for the disease. Patients arrived at the diagnostic laboratory of hereditary colorectal tumors (U.O.C. Clinical Molecular Biology) of the Federico II/CEINGE, University Hospital of Naples, between 2017 and 2023, for molecular diagnosis. Three samples of peripheral blood were obtained from all patients. DNA extraction from peripheral blood lymphocytes was carried out on two of the three blood test tubes drawn from each patient in order to obtain two different DNA aliquots, as previously described [15].
A population of 20 healthy subjects was also included into the study. This control population was recruited from unaffected members of at-risk families with previously negative results for the presence of the specific pathogenetic variant. All control subjects were matched for age (all adults) and were processed using the same workflow applied to individuals with suspected or confirmed adenomatous polyposis, including the use of the same sequencing panel and the same data analysis pipeline to minimize bias.
2.2. Molecular Screening of Gastrointestinal Polyposis Syndrome
To perform the molecular analysis of familial gastrointestinal polyposis, we set up the workflow reported in Figure 1.
The DNA extracted from the proband’s peripheral blood was first analyzed, using the next-generation sequencing (NGS) technique, for genes involved in adenomatous polyposis and, when pertinent (patients n° 13 and 44), also for genes involved in hamartomatous polyposis. Two gene panels, one specific for adenomatous polyposis syndromes, including 6 genes (APC (NM_000038.6), AXIN2 (NM_004655.3), MUTYH (NM_001048174.2), NTHL1 (NM_002528.7), POLD1 (NM_002691.4), and POLE (NM_006231.4)) and another specific for hamartomatous polyposis, including 10 genes (AKT1 (NM_001382430.1), BMPR1A (NM_004329.3), CDH1 (NM_004360.5), ENG (NM_001114753.3), PIK3CA (NM_006218.4), PTEN (NM_000314.8), SDHB (NM_003000.3), SDHD (NM_003002.4), SMAD4 (NM_005359.6), and STK11/LKB1(NM_000455.5)), as previously described [15], were analyzed via the next-generation sequencing technique using the AmpliSeq Library PLUS for Illumina Kit (catalog ID: 20019101, Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. The pooled and barcoded libraries were subsequently sequenced using the MiSeq Sequencing System (Illumina, San Diego, CA, USA). Variant calling and analysis were performed using Base Space Sequence HUB/variant interpreter Software v7.41.0 (basespace.illumina.com, San Diego, CA, USA). The raw FASTQ files generated and/or analyzed during the current study are available on Mendeley data (De Rosa, Marina (2025), “Molecular screening of adenomatous gastrointestinal polyposis syndrome”, Mendeley Data, V1, doi: 10.17632/2wxnmhwkhm.1)
The interpretation of the identified variants was performed in accordance with ACMG guidelines [28] using Varsome free software v13.12.2 (varsome.com, Saphetor SA, Lausanne, Switzerland) ([29] and Franklin by Genoox free software v90.1 (franklin.genoox.com, Tel Aviv District, Israel). Reference databases for hereditary colorectal tumors, such as the InSight-group (www.insight-group.org) and ClinVar (www.ncbi.nlm.nih.gov/clinvar, accessed on 28 March 2025) databases, were also examined.
Pathogenic variants and/or variants of unknown pathogenic significance, identified via NGS, were confirmed through polymerase chain reaction (PCR) and Sanger sequencing performed on a second, independently extracted DNA sample, using the primer pairs previously reported for the APC, MUTYH, and STK11 genes [15,30], and primers reported in Table S1 for the other analyzed genes. The use of a second DNA aliquot is recommended in order to minimize the risk of technical artifacts, sample handling errors, or sample swaps. Finally, for the copy number variation (CNV) investigation, the Multiplex Ligation-dependent Probe Amplification (MLPA) assay was offered to subjects in which no pathogenic/likely pathogenic variants were identified, also given the high frequency of large deletions described in the APC gene.
2.3. Statistical Analysis
To investigate genotype–phenotype correlations, patients were classified into three groups based on the molecular findings: carriers of one or more pathogenic variants (MUT^+^), carriers of variants of uncertain significance (VUS), and individuals without any identified variants (MUT^−^). Three phenotypic variables were analyzed across mutation groups: age at disease onset (continuous), polyp burden (ordinal), and inheritance pattern, assessed either as a binary Mendelian versus non-Mendelian classification or further subdivided into inheritance categories (e.g., DOMINANT or RECESSIVE). Inheritance patterns were determined based on pedigree analysis and clinical family history. Age at disease onset was treated as a continuous variable. Normality within groups was assessed using the Shapiro–Wilk test, and homogeneity of variances was evaluated using Levene’s test. Group comparisons were performed using one-way analysis of variance (ANOVA), followed by Holm-adjusted pairwise t-tests when appropriate. Because the reported number of gastrointestinal polyps included approximate values (e.g., “<10” and “>100”) and non-numeric clinical annotations, polyp burden was categorized into clinically meaningful ordered intervals (0, <10, 10–20, 20–50, 50–100, >100, and >1000) and analyzed as an ordinal variable. Group differences in polyp burden were therefore evaluated using the Kruskal–Wallis rank-sum test, followed by Dunn’s post hoc test with Holm correction for multiple comparisons. Categorical variables were analyzed using Pearson’s Chi-squared test applied to contingency tables. When overall significance was detected, post hoc pairwise comparisons of proportions were conducted with Holm-adjusted p-values. Heatmaps were used to visualize both adjusted p-values and group-wise count distributions. All statistical tests were two-sided, and a p-value < 0.05 was considered statistically significant. Correction for multiple testing was consistently applied using the Holm method across all pairwise comparisons.
In addition to p-values, effect size measures were reported to support clinical interpretability. For one-way ANOVA, eta-squared (η^2^) was calculated to quantify the proportion of variance in age at onset explained by mutation group. For Kruskal–Wallis analyses of ordinal polyp burden, epsilon-squared (ε^2^) was computed as a non-parametric measure of effect size (ε^2^ = (H − k + 1)/(n − k), where H is the Kruskal–Wallis statistic, k is the number of groups, and n is the sample size). For Chi-squared tests assessing associations between mutation group and inheritance pattern, Cramér’s V was estimated. Effect sizes were interpreted according to conventional thresholds (small, medium, and large).
All statistical analyses and visualizations were performed using R (version 4.4.2), with functions from the base stats package and the car, FSA, effectsize, ggplot2, ggstatsplot, dplyr, tidyr, and tibble packages.
2.4. Bioinformatic Analysis
To further investigate if the benign/likely benign variants identified could play any role in disease onset, a heatmap was generated using Microsoft Excel software that plotted each variant identified during the screening of the adenomatous gene panel, for each of the 80 subjects analyzed for the purpose of this study, reporting the presence of the variant in red and its absence in blue.
A second variant map was obtained by subtracting all variants identified in the healthy population from variants identified in affected subjects analyzed into the study. Afterward, to investigate a possible deleterious effect of these benign/likely benign variants, they were analyzed with the following software: Human Splicing Finder and UMD-Predictor Pro from Genomics https://genomnis.com (accessed on 28 March 2025) and HaploReg v4.2 (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php, accessed on 28 March 2025), using default settings. Analyses from these three software were performed separately and the results were successively combined to obtain a consensus framework. This integrative approach supported the selection of candidate variants for future functional validation.
Human Splicing Finder (HSF) is a bioinformatics tool designed to analyze DNA or RNA sequences to predict the impact of mutations on the pre-mRNA splicing process.
It provides a predictive score to estimate the probability that a variant alters physiological splicing. The significance thresholds for each change are the following:
Splice site score → change ≥10% → possible functional alteration
Splice site creation → score > 65–70 → potentially active
ESE/ESS ratio → alteration ≥2 → possible effect on splicing
ESE/ESS ratio → alteration ≥4 → likely relevant effect
UMD-Predictor Pro software evaluates the pathogenicity of a DNA variant using a combinatorial algorithm that integrates several criteria.
The algorithm calculates an overall normalized score on a scale from 0 to 100. Based on this score, variants are classified as follows:
- Polymorphism (likely benign) → Score < 50;
- Likely polymorphism → Score 50–64;
- Likely pathogenic mutation → Score 65–74;
- Pathogenic mutation → Score > 74 [31].
HaploReg v4.2 is a bioinformatics software designed for the functional analysis of genetic variants, mainly SNPs (single nucleotide polymorphisms).
The main functionalities are to provide information on histone modifications, chromatin state, transcription factor binding regions (TFBS), and DNase hypersensitivity, integrating data from projects such as ENCODE and Roadmap Epigenomics. This software also extends indexed SNPs to their SNPs in linkage disequilibrium (LD), using data from the 1000 Genomes Project for different populations, identifies potential regulatory effects of SNPs on target genes, such as expression quantitative trait loci (eQTLs), and assesses how a variant may alter transcription factor binding motifs, using motif databases such as TRANSFAC or JASPAR [32].
Together, these tools offer a comprehensive assessment of possible splicing, functional, and regulatory consequences, allowing a more complete interpretation of each variant’s potential impact.
Additionally, STRING analysis was performed to analyze the interactions between the following genes involved in the onset of gastrointestinal polyposis: APC, MUTYH, AKT1, AXIN2, STK11, POLD1, POLE, and NTHL. A STRING analysis is a bioinformatic approach that uses the STRING database to explore protein–protein interaction (PPI) networks. By inputting a list of genes or proteins, STRING identifies known and predicted interactions and visualizes them as a network [33,34].
3. Results
3.1. Main Findings
We established a molecular screening workflow for adenomatous gastrointestinal polyposis based on NGS analysis of a targeted gene panel, including APC, MUTYH, POLE, NTHL1, AXIN2, and POLD1. The selection of genes included in the multigene panel for adenomatous polyposis was based on the recommendations of the NCCN (National Comprehensive Cancer Network) [35], the ESMO (European Society of Medical Oncology) [36], the JSCCR (Japanese society for cancer of the colon and rectum) [37], and the ACMG (American College of Medical Genetics and Genomics) [38].
CNV analysis for APC and MUTYH genes was offered to patients without point pathogenic variants using the MLPA method.
When a differential diagnosis between adenomatous and hamartomatous polyposis could not be clearly established, patients were additionally analyzed using a specific panel targeting hamartomatous syndromes, including PTEN, STK11, SDHB, SDHD, BMPR1A, CDH1, AKT1, SMAD4, PI3KCA, and ENG genes. As suggested [39,40], probands who developed fewer than 10 adenomatous polyps but exhibited clinical features consistent with Lynch or Lynch-like syndromes were referred for MMR gene screening and excluded from this study.
As expected, the highest frequency of pathogenic variants was detected in APC and MUTYH genes, consistent with literature data [41,42]. By contrast, VUS were identified in a broader range of genes—APC, MUTYH, NTHL1, AXIN2, POLE, POLD1 and AKT1—with a relatively uniform distribution. This may reflect the limited functional characterization of VUS to date, as well as the fact that most prior screening efforts in adenomatous polyposis patients have focused primarily on APC and MUTYH, without the inclusion of other relevant genes.
The interpretation of VUS in our study was still largely based on computational predictions and was not complemented by solid functional data; experimental laboratory assays will be necessary to establish their pathogenic relevance more reliably.
The statistical analysis of clinical and molecular data, reported in the Methods section, revealed genotype–phenotype correlations among the three groups (MUT^+^, VUS, and MUT^−^) into which the patient cohort was stratified.
Specifically, MUT^+^ patients exhibited an earlier age of onset and significantly higher polyp counts compared to both VUS and MUT^−^ patients.
VUS carriers also differed significantly from MUT- patients in terms of the frequency of Mendelian inheritance, defined on the basis of pedigree analysis and clinical family history, which was even higher than that observed in MUT^+^ patients (0.82 vs. 0.55), whereas MUT^−^ patients showed a markedly low frequency (0.063).
Although this study is limited by the relatively small sample size, mainly the limited control group, and restricted gene panel analyzed, two key findings emerged from the analysis of benign variants:
-
Patients n° 18, 25, 26, 31, and 42 harbored a cluster of variants in POLD1, some of which were reported to be in LD, and bioinformatic analysis suggested a strong potential to cause splicing alterations. At least one SNP in each LD group was predicted to affect splicing. Furthermore, these variants localized to active regulatory regions and may alter transcription factor binding, also raising the possibility of altered expression of neighboring genes. On the positive strand, downstream of POLD1, lies MYBPC2, SPIB, and EMC10; upstream on the negative strand, is NAPSA. MYBPC2 encodes a myosin-binding protein involved in cardiomyopathies; SPIB encodes a lymphoid-specific transcription factor; EMC10 promotes angiogenesis and endothelial proliferation; and NAPSA encodes a protease inhibitor proposed as a marker in lung and renal cancers.
-
Patients n° 18, 25, 26, 53, and 55 carried the rs78429131 (APC: c.-31T > G) variant, located in the APC gene promoter. Bioinformatic evidence supported its potential role in modulating APC gene expression.
In this context, the possible crosstalk between gastrointestinal polyposis predisposing genes and molecular pathways, such as the BER system, Wnt signaling, and apoptosis, which contribute to shared cellular function, was investigated and is visually summarized in Figure 2, highlighting interactions among genes implicated in polyposis phenotypes.
Sixty patients were analyzed for adenomatous polyposis using genetic tests, as described in the Methods section.
We identified 33 patients (55%) carrying pathogenic variants in one of the analyzed genes, 11 patients (18.3%) harboring one or more variants of uncertain significance (VUS), and 16 patients (26.7%) were not informative, as they presented neither pathogenic/likely pathogenic nor VUS variants.
3.2. Genetic Findings (MUT+ Patients)
Among the 33 MUT^+^ patients, pathogenic variants were detected in the following genes: 21 in the APC gene, 1 in the AXIN2 gene, 10 in the MUTYH gene (with 17 distinct variants), and 1 in the NTHL1 gene, as reported in Table 1.
In total, the 40 identified pathogenic variants included:
- Eleven frameshift and indel variants (27.5%);
- Nine nonsense variants (22.5%);
- Four splicing variants (10%);
- Nine missense variants (22.5%);
- Four in-frame deletion variants (10%);
- Three large deletions (7.5%).
3.3. Genotype–Phenotype Correlations (MUT+ Patients)
Among the 21 patients carrying an APC mutation, 11 exhibited an autosomal dominant inheritance pattern, while the remaining 10 showed no evidence of Mendelian transmission. Two of these were confirmed as de novo cases (Table 2, patients n° 4 and 12), while for the other 8 cases, de novo origin could not be confirmed because their apparently healthy parents declined molecular screening (Table 2, patients n° 1, 2, 5, 7–9, 11, 15).
Of the 10 patients carrying mutations in the MUTYH gene, 5 exhibited a recessive pattern of inheritance, as expected, while the remaining 5 showed no clear evidence of Mendelian transmission. Three patients were found to carry a homozygous pathogenic variant in MUTYH (Table 2, patients n° 28–30), but only one of them had documented parental consanguinity (Table 2, patient n° 29).
The average age at disease onset in MUT^+^ patients was approximately 41 years, and the average number of polyps exceeded 100. Approximately 54% of these patients showed a Mendelian pattern of inheritance, as reported in Table 2.
A detailed molecular and clinical description is provided in Appendix A.
3.4. Genetic Findings (VUS Patients)
Among patients carrying only VUS, 3 carried variants in the APC gene, 1 in the MUTYH gene, 2 in the NTHL1 gene, 2 in the AXIN2 gene, 1 in the POLD1 gene, 1 in the POLE gene, and 1 in the AKT1 gene.
All VUS variants were missense, except for one splicing variant in the AKT gene (Table 3, patient n° 44).
3.5. Genotype-Phenotype Correlations (VUS Patients)
In this group of patients, the average age of onset was approximately 53 years; the average number of polyps was about 10, and approximately 82% of patients had a Mendelian type of inheritance, as reported in Table 3.
A detailed description of the clinical features of these patients, along with the criteria used for clinical classification of each variant, is provided in Appendix B.
3.6. Genetic Findings (MUT− Patients)
Sixteen patients were found to be not informative for the presence of either pathogenic variants or VUS in the analyzed gene panel.
3.7. Genotype-Phenotype Correlations (MUT− Patients)
Among the MUT^−^ patients, the average age at disease onset was approximately 53 years; the average number of polyps was around 10, and only approximately 6% of patients exhibited a Mendelian pattern of inheritance, as reported in Table 4.
All patients presented with more than 10 adenomatous polyps, except for patient n° 52, who developed only 4 polyps.
3.8. Statistical Analysis Results
From the statistical analysis, performed as described in the Methods section, differences between the three groups of patients emerged. For age at onset (Figure 3A), MUT^+^ patients were significatively younger than both VUS and MUT^−^ patients (mean values of ~40, ~53, and ~55 years, respectively). The one-way ANOVA showed a significant group effect, and the associated effect size was large (η^2^ ≈ 0.23; 95% CI: 0.02–0.42), indicating that a substantial proportion of the variance in age at onset was explained by the mutation class.
For the number of gastrointestinal adenomatous polyps (Figure 3B), MUT^+^ patients developed a much higher polyp burden (>100 polyps) compared to VUS and MUT^−^ patients (>10 polyps). Polyp burden differed significantly among mutation groups (Kruskal–Wallis test, χ^2^(2) = 23.53, p = 7.76 × 10^−6^), with a large effect size (ε^2^ = 0.38), indicating that mutation class explained a substantial proportion of the variability in polyp burden.
Regarding Mendelian inheritance (Figure 3C), the three groups did not behave in a dependent manner (Chi-squared = 17.415, df = 2, p = 0.0001654). The effect size for this association was large (Cramér’s V ≈ 0.50; 95% CI ≈ 0.25–0.68), indicating that inheritance patterns strongly differed depending on mutation status. Specifically, only ~6% of MUT^−^ patients (1/16) showed a Mendelian inheritance pattern. By contrast, ~54.5% of MUT^+^ patients (18/33) displayed Mendelian transmission: 36.4% (12/33) dominant, 15% (5/33) recessive, and 3% (1/33) dominant for tumors. Among VUS patients, approximately 82% (9/11) exhibited dominant Mendelian inheritance; notably, 5 of the 9 patients had a gastrointestinal polyposis phenotype, and 4 had gastrointestinal tumors (Figure 3D) (Chi-squared = 29.873, df = 6, p = 4.156 × 10^−5^).
Effect size for categorical binary outcomes (Mendelian inheritance TRUE/FALSE) was quantified using Cramér’s V. For the contingency table with multiple inheritance subclasses, post hoc standardized residuals and Holm-adjusted p-values were used instead of global effect size, as recommended for sparse multi-category tables.
3.9. Bioinformatic Analysis Findings
We considered all benign or likely benign variants identified in the 80 subjects analyzed in this study and generated a heatmap illustrating the presence (red squares) or absence (blue squares) of these variants in each gene across subjects (Figure S1). This analysis was performed to understand whether benign or likely benign variants occurring in the same gene or gene network might have a cumulative impact on disease onset and/or phenotypic manifestations.
Subsequently, variants observed in the unaffected control population were subtracted from those identified in patients, resulting in a refined mutational heatmap that displayed, for each patient, all variants not detected in healthy control individuals and their corresponding minor allele frequencies (MAFs) (Figure 4).
As shown in Figure 4, the majority of benign variants of affected subjects showed low MAF, with 58% of the variants displaying MAF <1%, 13% between 1% and 2%, and 29% between 2% and 11%. By contrast, healthy subjects exhibited a predominance of common variants, with 62% of subjects showing MAF >10%, 18% between 2% and 9%, while only 20% presented MAF < 1%.
Interestingly, no benign variants of the NTHL1 gene were observed in healthy subjects and MAFs of benign variants identified in affected subjects was <0.1%.
This distribution supported a potential partially disruptive effect of benign variants in accordance with differences in allele frequency. Furthermore, the absence of benign variants in the NTHL1 gene could indicate that the gene was functionally essential and therefore the variants assumed a pathogenic significance with high probability.
Each benign variant was analyzed using bioinformatic tools, as described in the Methods section.
Analysis of the refined heatmap revealed, in our opinion, three noteworthy observations, consisting of the identification of:
- A cluster of benign variants in the POLD1 gene in patients n° 18, 31, and 42.
- The rs78429131 variant of the APC promoter region, named APC: c.-31T > G, in patients n° 18, 25, 26, 53, and 55.
- Other benign variants identified in MUT^−^ patients were suggested to be partially disruptive.
The POLD1 cluster variants (point 1) comprising the SNPs are listed in Table 5.
Results from HaploReg v4.2 analysis (a software examining functional consequences of SNPs on gene expression) of this cluster of variants identified two groups of SNPs in linkage disequilibrium (as reported in the Table 5), including the following: rs1726804, rs3212328, rs1143666, rs3218764, and rs1274607 (the first group), and rs3212330, rs2463239, rs2463238, and rs112856489 (the second group), all of which showed an r^2^ (coefficient of determination) ≥ 0.8.
In addition, analysis with the Human Splicing Finder (HSF) tool (a software examining the potential deleterious effect on SNPs on splicing mechanism) predicted significant alterations for SNPs rs3219384, rs1274607, and rs112856489.
rs3219384 and rs1274607 (the latter being the only SNP in this cluster not in linkage disequilibrium with other SNPs) were predicted to significantly alter the enhancer/silencer ratio (ESE/ESS) by approximately +4-fold and −8-fold, respectively. These changes could result in exon skipping or intron retention events.rs112856489 was predicted to disrupt a wild-type acceptor splice site, with a decrease in splicing score of −33.09% (from 77.75 to 52.02), indicating a likely functional impact.
HaploReg v4.2 analysis also revealed that each of these polymorphisms mapped to DNase I hypersensitive sites (DHS)—regions of DNA devoid of nucleosomes and thus accessible to transcription factors. Furthermore, all SNPs overlapped with genomic regions marked by histone modifications characteristic of active enhancers or promoters, in a tissue-specific manner, as detailed in Table 6.
These findings suggested that the identified variants were located within putative regulatory regions, potentially modulating gene expression in specific tissues.
This hypothesis was supported by the observation that these SNPs can create or disrupt transcription factor binding motifs or alter the binding affinity of transcription factors for their target DNA sequences.
On the negative DNA strand, the variants:
- ○Create recognition sites for E2F, PU.1, SRF, Sin3A, TATA-box, CAC-binding protein, Egr-1, Ets, and SP1;
- ○Disrupt sites for Sin3A, BCL, and ZBTB7A;
- ○Increase affinity for SP1 and STAT binding motifs. On the positive DNA strand, the variants:
- ○Create recognition sites for p300 and RXRA;
- ○Increase affinity for motifs bound by GLI, NF-κB, NRSF, and CCNT2.
Furthermore, SNP rs1274607 mapped to a highly conserved genomic region. Several of the analyzed SNPs were located within specific DNA-protein binding sites, including:
- rs1726804 within a ZNF263 binding site.
- rs3212330, rs2463239, and rs2463238 within a POL2 binding site.
- rs3212330, also within a ZEB1 binding site.
The rs78429131 variant (APC: c.-31T > G) (point 2), located at position chr5-112043384 (GRCh37), was observed in patients n° 18, 25, 26, 53, and 55. It showed a population frequency of 0% according to the GnomAD database.
HaploReg v4.2 analysis suggested a possible deleterious effect on gene/protein expression of this SNP. This variant was indeed classified as an expression quantitative trait loci (eQTL) and was located within a Dnase I hypersensitive site, overlapping histone modifications associated with active promoter regions (TSSA; PROM_D1) and regulatory elements such as H3k4me1_Enh, H3K4me3_Pro, H3K27ac_Enh, and H3K9ac_Pro, all in a tissue-specific manner.
These chromatin marks are particularly enriched in gastrointestinal tissues, including:
- Colon mucosa and smooth muscle;
- Duodenal mucosa and smooth muscle;
- Esophagus;
- Rectal mucosa and smooth muscle;
- Sigmoid colon;
- Small intestine.
The variant also overlapped with binding sites for several transcription factors, including POL2, POL24H8, SIN3A, OCT2, POU2F2, and NFKB. Importantly, the presence of the rs78429131 polymorphism created a de novo binding site for the transcription factor HMX1, a transcription factor belonging to the H6 family of homeobox proteins, which often acts as a transcriptional repressor of genes involved in the developmental morphogenesis of the eye and specific nervous system structures.
Unfortunately, due to the lack of RNA and protein extracts from these patients, we were unable to perform additional molecular analyses to assess the functional consequences of these variants.
Finally, since MUT^−^ patients appeared to represent a sub-population with familial predisposition to the disease, rather than individuals affected by a Mendelian disorder, we conducted a bioinformatic analysis on benign variants identified in these patients that were absent in healthy controls (point 3). As a result of this analysis, we highlight the following notable findings:
- Patient n° 48 carried the POLE variant c.2174-8G > A, which was predicted using HSF (Human Splicing Finder) software to potentially alter splicing by activating a cryptic splice donor site, with a score variation of 18.56% (from 55.45 to 65.74).
- Patient n° 51 harbored two POLE variants classified as benign: c.6494G > A and c.330 + 66G > A. Both were predicted to cause splicing alterations by HSF.
- ○The first variant would significantly alter the ESE/ESS motif ratio (−2);
- ○The second would activate a cryptic splice site, with a score variation of 53.64% (from 51.96 to 79.83);
- ○Moreover, POLE c.6494G > A was classified as probably pathogenic by the UMD predictor, with a score of 67.
- Patient n° 53 carried the POLD1 variant c.1893-60G > A, which was predicted to cause activation of a cryptic splice acceptor site, with a score variation of 73.71% (from 37.81 to 65.68) according to HSF.
- Patient n° 55 carried the MUTYH variant c.304 + 56G > A, also found in patient n° 43, which was predicted to activate a cryptic splice acceptor site, with a score variation of 64.6% (from 43.14 to 71.01) by HSF.This patient also carried the rs78429131 variant (APC: c.-31T > G), as previously discussed.
- Patient n° 60 carried the POLE variant c.91G > T, also found in patient n° 39 (GnomAD frequency: 1.22%), and predicted by HSF to be potentially deleterious and to activate a cryptic donor site, with a score variation of 71.03% (from 38.21 to 65.35).
All of these variants were suggested to disrupt gene expression through a mechanism involving splicing alterations. Of these patients, only patient n° 53 showed evidence of Mendelian inheritance.
4. Discussion
In agreement with the results of molecular and statistical analyses, we propose that the VUS identified in this study may contribute to disease onset, likely resulting in a milder phenotype but still following a Mendelian inheritance pattern. Conversely, MUT^−^ patients exhibit phenotypic similarities to VUS carriers but have a much lower incidence of Mendelian inheritance.
Effect size estimates derived from our statistical analyses further reinforced these observations. The large effect sizes for age at onset (η^2^ ≈ 0.23) and polyp burden (ε^2^ ≈ 0.38) indicated that mutation class accounts for a substantial proportion of phenotypic variability. Likewise, the strong association between mutation status and Mendelian inheritance (Cramér’s V ≈ 0.50) demonstrated that inheritance patterns differ markedly across patient subgroups. Together, these measures complement traditional significance testing and support the interpretation that MUT^+^, VUS, and MUT^−^ individuals represent biologically distinct categories.
This supports the hypothesis that MUT- patients represent cases with familial predisposition or sporadic polyposis, rather than true Mendelian disease. It is likely that these patients do not carry any single variant sufficient to cause disease, but the disease phenotype is the result of additive effects of multiple partially disrupting variants.
Based on the results obtained from the bioinformatic analyses, we hypothesize a possible contribution of the cluster variant polymorphisms in the POLD1 gene and other benign variants in the APC gene (the rs78429131 variant; APC: c.-31T > G), but also in the POLE and MUTYH genes, to disease onset, progression, or phenotype, potentially through additive effects involving multiple molecular mechanisms. To confirm this hypothesis, further studies analyzing the mRNA and protein expression of these genes, as well as investigating the additive or synergistic effects of co-occurring benign or VUS variants through a systems biology approach, are needed.
The pathogenicity of monoallelic MUTYH and NTHL1 variants remains controversial. Monoallelic germline MUTYH mutations have long been known to increase lifetime cancer risk [43,44,45]. However, risk estimates for monoallelic carriers remain variable across populations and study designs, suggesting the influence of additional genetic or environmental modifiers.
While earlier reports suggested that monoallelic NTHL1 variants were not associated with increased tumor or polyposis risk [46,47], more recent evidence shows that tumors with biallelic and monoallelic NTHL1 mutations share somatic mutational patterns [48,49], and monoallelic NTHL1 variants may elevate lifetime cancer risk [46,50]. It is conceivable that, in some patients, pathogenic phenotypes arise from the additive effects of monoallelic variants in recessive genes such as MUTYH and NTHL1, combined with additional mildly deleterious variants in the same or other genes.
Finally, we hypothesize that the MUT^−^ patient group may represent cases in which disease onset results from a cumulative burden of multiple partially deleterious variants in crosstalking predisposing genes and interacting molecular pathways that contribute to shared cellular function.
In conclusion, our findings support distinct etiologies between MUT^+^ and VUS patients compared to MUT^−^ individuals. We propose that benign variants could represent mildly deleterious variants, that either singly or in combination, may act as disease modifiers, contributing to polyposis risk via additive effects across shared molecular pathways. Future studies with expanded gene panels and larger cohorts, including functional analysis of genomic variants, are needed to validate these hypotheses.
In light of these observations, this work presents an important innovative contribution and suggests that the interpretation of gene variants cannot be fully understood by the pathogenic/benign dichotomy. Our results support a model in which benign variants in specific genes, such as POLD1 and APC, are not biologically irrelevant but can nevertheless have a disruptive impact on the gene or protein of variable entity. These variants, although not able to cause the disease phenotype by themselves, can determine quantitative modulations of gene expression or protein activity. The disease phenotype could result from the combined effects of multiple variants in the same gene or in genes of the same molecular pathway or crosstalking pathways. In this context, these variants could modulate the expression of the phenotype, such as disease penetrance or disease severity, expressed in terms of age of onset and number of polyps developed by patients. Our findings also highlight the need to consider the effect that variants have on the regulation of gene expression, and not just on protein function, as programmed in the main tools that define the classification of gene variants. Recognizing their potential roles as modifiers of certain variants could improve phenotype prediction and risk stratification.
Future Perspective
The results obtained from this study open up interesting future perspectives for translational research. First of all, it will be necessary to confirm the etiological distinction of the disease between MUT^+^, MUT^−^, and VUS patients through multicenter studies. Simultaneously, it will be necessary to enlarge the gene panels screened in patients with polyposis or consider a whole-genome approach. This will potentially allow the identification of new genes responsible for the onset of disease, as well as the cumulative contribution of benign variants. In vitro and in vivo studies in cellular and animal models will be necessary for a functional analysis of VUS variants and those classified as benign variants. Finally, polygenic risk models should be considered to assess the additive or synergistic role of partially deleterious variants. A better understanding of the etiology of the disease, as well as the role that only partially destructive variants may play, could influence diagnostic interpretation, risk stratification, and personalized surveillance strategies in patients with polyposis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Olkinuora A.P. Peltomäki P.T. Aaltonen L.A. Rajamäki K. From APC to the genetics of hereditary and familial colon cancer syndromes Hum. Mol. Genet.202130 R 206R 22410.1093/hmg/ddab 20834329396 PMC 8490010 · doi ↗ · pubmed ↗
- 2De Rosa M. Rega D. Costabile V. Duraturo F. Niglio A. Izzo P. Pace U. Delrio P. The biological complexity of colorectal cancer: Insights into biomarkers for early detection and personalized care Therap. Adv. Gastroenterol.2016986188610.1177/1756283 X 1665979027803741 PMC 5076770 · doi ↗ · pubmed ↗
- 3Turano M. Delrio P. Rega D. Cammarota F. Polverino A. Duraturo F. Izzo P. De Rosa M. Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy Cancers 201911193210.3390/cancers 1112193231817090 PMC 6966638 · doi ↗ · pubmed ↗
- 4De Rosa M. Pace U. Rega D. Costabile V. Duraturo F. Izzo P. Delrio P. Genetics, diagnosis and management of colorectal cancer (Review)Oncol. Rep.2015341087109610.3892/or.2015.410826151224 PMC 4530899 · doi ↗ · pubmed ↗
- 5Talseth-Palmer B.A. The genetic basis of colonic adenomatous polyposis syndromes Hered. Cancer Clin. Pract.201715510.1186/s 13053-017-0065-x 28331556 PMC 5353802 · doi ↗ · pubmed ↗
- 6Borun P. De Rosa M. Nedoszytko B. Walkowiak J. Plawski A. Specific Alu elements involved in a significant percentage of copy number variations of the STK 11 gene in patients with Peutz-Jeghers syndrome Fam. Cancer 20151445546110.1007/s 10689-015-9800-525841653 PMC 4559094 · doi ↗ · pubmed ↗
- 7De Rosa M. Galatola M. Quaglietta L. Miele E. De Palma G. Rossi G.B. Staiano A. Izzo P. Alu-mediated genomic deletion of the serine/threonine protein kinase 11 (STK 11) gene in Peutz-Jeghers syndrome Gastroenterology 20101382558256010.1053/j.gastro.2010.03.06120435009 · doi ↗ · pubmed ↗
- 8Yehia L. Heald B. Eng C. Clinical Spectrum and Science Behind the Hamartomatous Polyposis Syndromes Gastroenterology 202316480081110.1053/j.gastro.2023.01.02636717037 · doi ↗ · pubmed ↗