NF-κB Signaling as a Central Driver of Cancer Cachexia
Yan Li, Hao Jiang, Rui Chen, Haitao Huang, Shengguang Ding

TL;DR
This paper explores how the NF-κB signaling pathway drives cancer cachexia, a condition causing severe muscle and fat loss, and suggests targeting this pathway could lead to better treatments.
Contribution
The paper identifies NF-κB as a central driver of cancer cachexia and proposes it as a viable therapeutic target.
Findings
NF-κB signaling promotes muscle wasting, adipose tissue disruption, and metabolic imbalance in cancer cachexia.
Pharmacologic and nutritional interventions targeting NF-κB show promise in reducing cachexia symptoms.
Combining NF-κB modulation with other therapies may offer synergistic benefits for treating cancer cachexia.
Abstract
Cancer cachexia is a debilitating complication that profoundly affects patient quality of life and treatment outcomes, yet its underlying mechanisms remain incompletely understood. Accumulating evidence indicates that persistent inflammation plays a central role in driving muscle wasting, adipose tissue remodeling, and systemic metabolic imbalance. This article provides an integrated overview of how inflammatory signaling coordinates pathological changes across multiple organs, highlighting its contribution to the progression of cachexia. By synthesizing current mechanistic insights, this work aims to clarify key regulatory pathways and identify unifying concepts that advance understanding of disease pathogenesis. These insights may help guide future research and support the development of more effective, mechanism-based therapeutic strategies for cancer cachexia. Cancer cachexia is a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
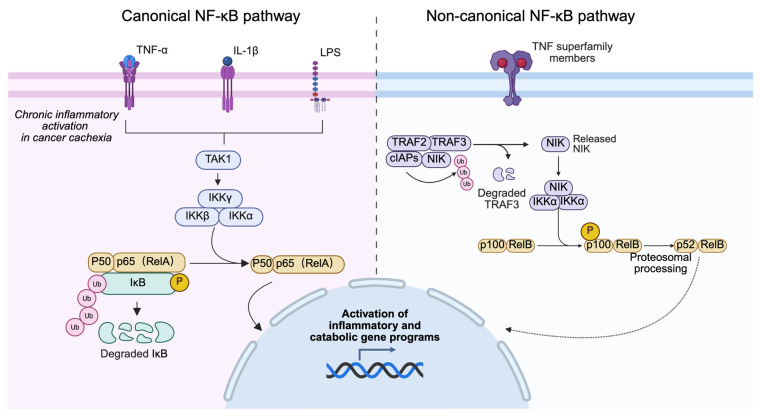 Figure 1
Figure 1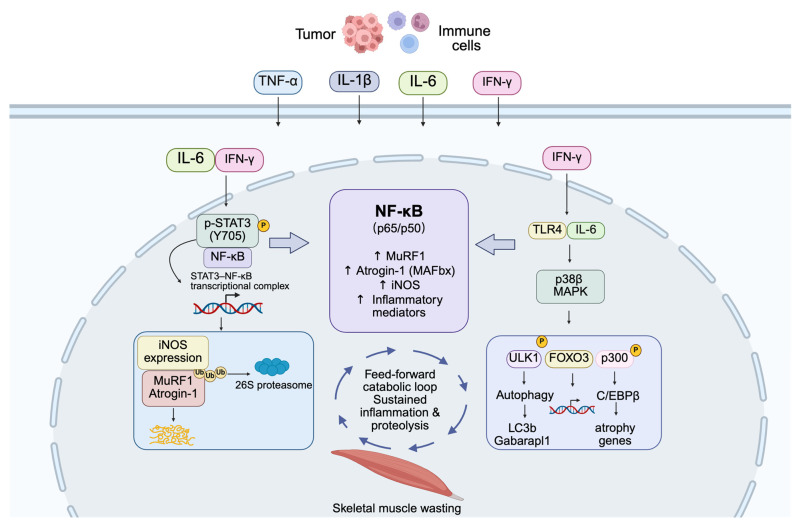 Figure 2
Figure 2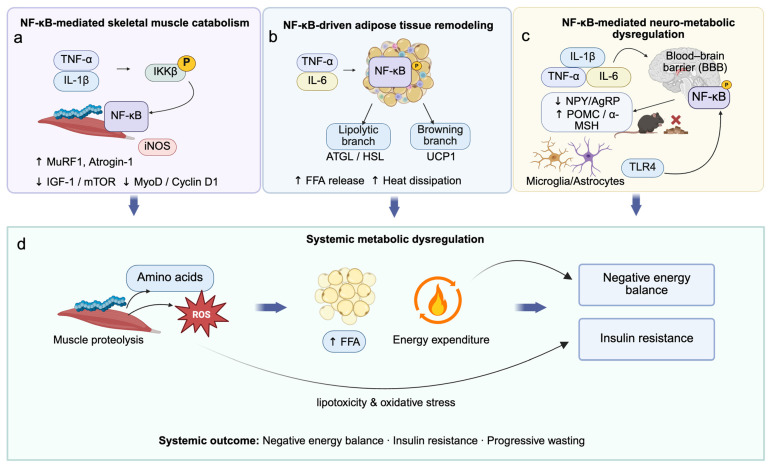 Figure 3
Figure 3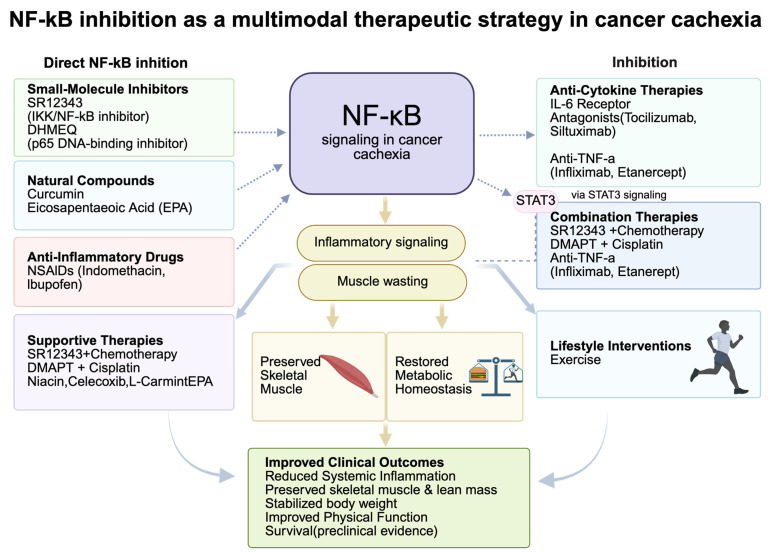 Figure 4
Figure 4- —Nantong First People’s Hospital Provincial and Ministerial Level High-level Science and Technology Project Cultivation Fund
- —Nantong Municipal Health Commission Scientific Research Project (Directive)
- —Nantong Municipal Health Commission
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMuscle Physiology and Disorders · Nutrition and Health in Aging · GDF15 and Related Biomarkers
1. Introduction
Cancer cachexia is a multifactorial metabolic syndrome characterized by progressive skeletal muscle wasting, unintentional weight loss, and profound systemic energy imbalance. Importantly, cancer cachexia is more than malnutrition; it represents a hypermetabolic, paraneoplastic syndrome that is largely unresponsive to conventional nutritional support. It is driven by chronic inflammation, enhanced proteolysis, and impaired tissue regeneration, and it is strongly associated with poor treatment tolerance, reduced quality of life, and increased mortality [1,2]. In addition to muscle wasting, cancer cachexia is characterized by maladapted adipose tissue metabolism, including loss of white adipose tissue and pathological browning, together with systemic inflammation marked by elevated TNF-α, IL-6, IL-1β, and related cytokines. Among the numerous molecular drivers implicated in cachexia, the nuclear factor κB (NF-κB) signaling pathway has emerged as a central orchestrator of disease pathophysiology [3]. These features collectively define a cachectic cascade in which inflammatory and metabolic alterations reinforce one another. Pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β), secreted by tumors and immune cells, activate the IκB kinase (IKK) complex, leading to phosphorylation and degradation of IκB inhibitors. [4,5]. This process permits nuclear translocation of NF-κB dimers predominantly p50/RelA—which induce transcription of key catabolic genes, including MuRF1, Atrogin-1, and iNOS [3]. Consequently, both the ubiquitin–proteasome system (UPS) and the autophagy–lysosome pathway (ALP) are activated, accelerating myofibrillar protein degradation [6]. In parallel, TNF-α suppresses the IGF-1/Akt/mTOR anabolic signaling cascade, further exacerbating the imbalance between protein synthesis and degradation [7,8]. Together, these processes position NF-κB as a critical molecular link between inflammation and muscle atrophy in cancer cachexia.
Beyond its role in proteolysis, sustained NF-κB activation profoundly disrupts muscle regeneration and tissue homeostasis. NF-κB upregulates Pax7 in satellite cells while repressing myogenic regulators such as Cyclin D1 and MyoD, thereby impairing myoblast differentiation and regeneration of damaged fibers [9]. Importantly, NF-κB signaling integrates multiple components of the cachectic cascade across different cellular compartments. NF-κB activation occurs across multiple cellular compartments including myofibers, fibro-adipogenic progenitors (FAPs), and infiltrating macrophages forming a multicellular inflammatory feedback loop that perpetuates local tissue degeneration [3]. Elevated reactive oxygen species (ROS) further amplify NF-κB signaling and cooperate with FOXO transcription factors to reinforce UPS and ALP mediated proteolysis while suppressing mTOR activity [10]. In addition, cytokine crosstalk particularly between IFN-γ and TNF-α engages the STAT3–NF-κB axis, promoting iNOS expression and intensifying catabolic signaling [11]. Collectively, this interconnected network of inflammation, oxidative stress, and regenerative failure underscores the pivotal role of NF-κB in skeletal muscle dysfunction during cachexia [12,13].
Mechanistically, NF-κB is a highly conserved intracellular signaling pathway that translates diverse extracellular cues including cytokines, pathogen-associated ligands, and oxidative stress into specific gene expression programs. As a central node of the cachexia cascade, NF-κB links inflammatory signaling to tissue-specific metabolic remodeling. The NF-κB family comprises five subunits (p50, p52, RelA/p65, RelB, and c-Rel), all of which share a Rel homology domain responsible for DNA binding and dimerization [14,15]. Canonical NF-κB signaling is activated by stimuli such as TNF-α, IL-1β, and Toll-like receptor ligands through TAK1-mediated phosphorylation of IKKβ within the IKK complex (IKKα, IKKβ, and NEMO/IKKγ), leading to IκBα degradation and nuclear translocation of NF-κB dimers [16]. In contrast, the non-canonical pathway relies on NF-κB-inducing kinase (NIK) and IKKα to promote p100 processing into p52 and nuclear accumulation of RelB-containing dimers, primarily regulating adaptive immune responses [17,18]. While transient NF-κB activation is essential for immune defense and tissue repair, chronic or dysregulated activation drives persistent inflammation, progressive muscle wasting, adipose tissue remodeling, and tumor progression [19]. Consistent with this, preclinical studies demonstrate that both genetic and pharmacological inhibition of NF-κB using IKK inhibitors (e.g., DHMEQ, SR12343), natural compounds such as curcumin, or low-dose nonsteroidal anti-inflammatory drugs (NSAIDs) can attenuate muscle wasting and preserve lean mass [20,21,22]. These findings establish NF-κB as a central molecular node in cancer cachexia and support its therapeutic targeting through multimodal strategies that integrate anti-inflammatory agents, metabolic modulation, and exercise-based rehabilitation. Accordingly, this review aims to delineate the multifaceted roles of NF-κB in cancer cachexia across skeletal muscle, adipose tissue, systemic metabolism, and central appetite regulation, and to discuss current and emerging therapeutic strategies targeting this pathway.
2. Overview of NF-κB Signaling Pathway
The NF-κB family comprises five evolutionarily conserved transcription factors p50 (processed from p105), p52 (from p100), RelA (p65), RelB, and c-Rel each containing a Rel Homology Domain (RHD) at the N-terminus that facilitates DNA binding, dimerization, and interaction with inhibitory IκB proteins [23,24,25]. Under basal conditions, NF-κB dimers are retained through association with IκB family members or precursor proteins containing ankyrin repeats. [26,27]. NF-κB signaling is classically divided into two major branches: the canonical and non-canonical pathways, which differ in activation triggers, signaling kinetics, and biological outputs [15,28].
In the canonical pathway, pro-inflammatory stimuli such as TNF-α, IL-1β, Toll-like receptor ligands, and chemotherapy-associated stress activate transforming growth factor-β–activated kinase 1 (TAK1), leading to phosphorylation of the IKK complex [28,29]. IKKβ-mediated degradation of IκBα permits rapid nuclear translocation of RelA/p50 heterodimers, inducing transcription of genes involved in inflammation, stress responses, and cellular metabolism [30]. Additionally, IKKβ phosphorylates p65 on Ser536, enhancing its transcriptional potency [31]. Functionally, this branch predominates in acute inflammatory responses and stress responses and, under conditions of sustained activation, contributes to pathological processes such as skeletal muscle atrophy in cancer cachexia through induction of MuRF1, Atrogin-1, and iNOS [3,9,32,33]. In contrast, the non-canonical NF-κB pathway is selectively activated by a restricted subset of tumor necrosis factor receptor superfamily members, including CD40, BAFF-R, and lymphotoxin-β receptor. This pathway depends on stabilization of NF-κB-inducing kinase (NIK) and IKKα-mediated processing of p100 into p52, generating p52/RelB heterodimers that regulate genes involved primarily in immune cell differentiation and lymphoid homeostasis [15,34]. Compared with canonical signaling, non-canonical activation is slower and relies on de novo protein synthesis. Importantly, current evidence supporting a dominant role for NF-κB signaling in cancer cachexia is derived predominantly from experimental models in which the canonical IKKβ–RelA axis is persistently activated. In widely used cachexia models including C26 colon carcinoma, Lewis lung carcinoma, and Apc^Min/+ mice canonical NF-κB signaling drives skeletal muscle proteolysis, adipose tissue remodeling, and systemic inflammation. In contrast, the contribution of non-canonical NF-κB signaling to cachexia appears to be context-dependent and remains incompletely defined, with available data suggesting a more indirect role in immune remodeling rather than direct induction of tissue wasting.
Despite their distinct upstream regulators, canonical and non-canonical pathways are interconnected through regulatory crosstalk that fine-tunes NF-κB activity [28,35]. However, in the context of cancer cachexia, sustained activation of the canonical pathway rather than balanced physiological signaling emerges as the principal mechanism linking chronic inflammation to metabolic and catabolic dysfunction [22]. Accordingly, subsequent sections focus on cachexia-specific NF-κB activation patterns and downstream pathological consequences across target tissues (Figure 1).
3. NF-κB Activation via Pro-Inflammatory Cytokines: Linking Inflammation to Muscle Wasting
3.1. Cytokine-Driven NF-κB Activation and Its Catabolic Impact in Cancer Cachexia
Cancer cachexia is characterized by chronic systemic inflammation, with elevated circulating levels of TNF-α, IL-1β, IL-6, and interferon-γ (IFN-γ) [36]. These cytokines converge on NF-κB signaling in skeletal muscle, establishing a pro-catabolic transcriptional program that disrupts muscle proteostasis [3]. In experimental cachexia models, TNF-α robustly activates the canonical IKKβ–NF-κB pathway, leading to upregulation of muscle-specific E3 ubiquitin ligases such as MuRF1 and Atrogin-1, thereby accelerating ubiquitin–proteasome-mediated protein degradation [37,38]. IL-1β further amplifies muscle catabolism through NF-κB-dependent induction of cyclooxygenase-2 and hypoxia-responsive pathways, while IL-6 activates the gp130/JAK/STAT3 axis and synergistically enhances NF-κB signaling [39,40,41]. IFN-γ cooperates with TNF-α to promote STAT3–NF-κB complex formation, driving iNOS expression and oxidative stress–associated proteolysis [42,43]. Together, these cytokines generate a feed-forward inflammatory loop that sustains NF-κB activation and progressive muscle wasting [14]. Notably, the majority of mechanistic evidence supporting these pathways originates from preclinical models, where pharmacologic or genetic inhibition of IKKβ or RelA consistently attenuates muscle loss. In contrast, human studies largely demonstrate correlations between circulating inflammatory cytokines, NF-κB target gene expression, and cachexia severity, underscoring a gap between mechanistic validation and clinical causality.
3.2. NF-κB as a Central Integrator of Systemic Inflammation in Cachexia
In cancer cachexia, NF-κB functions as a context dependent integrator of chronic inflammatory signals rather than a generic or uniformly acting inflammatory switch. At the core of these inflammatory responses lies NF-κB, a master transcriptional regulator that integrates extracellular inflammatory cues into gene expression programs governing immunity, metabolism, and tissue homeostasis [28,44]. In unstimulated cells, NF-κB dimers (primarily p65/p50) are sequestered in the cytoplasm by inhibitory IκB proteins [45]. Upon activation of Toll-like receptors (TLRs) or cytokine receptors (e.g., TNFR, IL-1R), the IKK complex phosphorylates IκB, marking it for proteasomal degradation and releasing NF-κB to translocate into the nucleus [46]. Once activated, NF-κB drives the expression of pro-inflammatory cytokines, chemokines, and adhesion molecules, thereby amplifying inflammatory cascades [47,48]. Importantly, the pathological relevance of NF-κB signaling in cancer cachexia arises from its sustained and cell type specific activation across multiple tissues. In macrophages, NF-κB upregulates TNF-α and IL-6, reinforcing systemic inflammation; in endothelial cells, it promotes leukocyte recruitment through induction of VCAM-1 and ICAM-1; and in adipocytes, TLR4 activation by free fatty acids engages NF-κB signaling, contributing to metabolic stress and insulin resistance [49,50,51]. A key regulatory feature of NF-κB is its self-reinforcing loop TNF-α induces NF-κB, which in turn promotes further TNF-α transcription, sustaining inflammation [52]. This persistent activation state enables NF-κB to integrate systemic inflammatory cues into coordinated transcriptional programs that promote tissue catabolism and metabolic dysregulation. Although targeting NF-κB signaling (e.g., via IKKβ inhibitors or p65 silencing) shows clear efficacy in experimental models, therapeutic translation remains challenging due to the risk of impairing essential immune and regenerative functions [53].
3.3. Synergistic Interactions Between NF-κB, STAT3, and p38 MAPK Signaling
In cachectic skeletal muscle, NF-κB activation cooperates with other catabolic pathways to exacerbate protein degradation [8,54]. One prominent axis involves STAT3, which is activated by IL-6, IFN-γ, and TNF-α. Upon phosphorylation (Y705), STAT3 forms a complex with NF-κB (p65), and together they translocate to the nucleus to functionally cooperate with NF-κB to enhance iNOS/NO catabolic signaling, triggering the iNOS/NO catabolic cascade [42]. Persistent STAT3 activation further reinforces muscle wasting by upregulating caspase-3 and muscle-specific E3 ubiquitin ligases, including MAFbx/Atrogin-1, thereby accelerating ubiquitin–proteasome system-mediated protein degradation [55]. Simultaneously, the β isoform of p38 MAPK activated downstream of TLR4 and inflammatory cytokine signaling phosphorylates p300 (Ser12), leading to activation of the transcription factor C/EBPβ and induction of muscle atrophy-related genes [55]. p38β MAPK also promotes autophagic by phosphorylating ULK1 (Ser555) and upregulates genes involved in lysosomal and proteasomal pathways, such as LC3b and Gabarapl1 [55]. Collectively, these pathways form a feed-forward catabolic signaling network in experimental models of cancer cachexia, in which NF-κB acts as a central coordinating node that integrates inflammatory and stress-responsive inputs rather than functioning as an isolated or exclusive driver. The cooperative activation of NF-κB, STAT3, and p38 MAPK signaling establishes a feed-forward catabolic program that sustains inflammation, proteolysis, and autophagic activation, ultimately driving progressive skeletal muscle wasting in cancer cachexia (Figure 2).
4. Pathological Mechanisms of NF-κB in Cancer Cachexia
4.1. NF-κB as a Central Regulator of Muscle and Adipose Catabolism in Cancer Cachexia
Cancer cachexia is marked by severe loss of skeletal muscle and adipose tissue, largely driven by persistent systemic inflammation and dysregulated catabolic signaling [56]. Central to this process is aberrant activation of the NF-κB pathway, which integrates inflammatory cues to regulate muscle proteolysis, impaired regeneration, and adipose remodeling [3,54]. In skeletal muscle, elevated levels of TNF-α, IL-1β, and IL-6 activate the IKKβ NF-κB axis, resulting in IκBα degradation and nuclear translocation of p65/p50 heterodimers [57]. These complexes induce expression of catabolic genes such as MuRF1, Atrogin-1, and iNOS, thereby promoting protein degradation through both the UPS and the ALP [6,9,58]. Simultaneously, NF-κB suppresses the IGF-1/Akt/mTOR anabolic axis and inhibits myogenic differentiation via Pax7 upregulation and repression of Cyclin D1 and MyoD [9]. These effects are reinforced by macrophage and fibro-adipogenic progenitor (FAP) infiltration, establishing a feed-forward loop of inflammation and tissue degradation. Muscle proteostasis is maintained by coordinated UPS and ALP activity; however, excessive activation of both pathways regulated in part by FOXO transcription factors exacerbates muscle wasting [59]. The concurrent upregulation of MuRF1 and Atrogin-1 is a hallmark of cachexia and a validated therapeutic target, with their inhibition shown to delay atrophy in preclinical models [60].
In white adipose tissue (WAT), NF-κB contributes to metabolic dysfunction by promoting lipolysis and white-to-brown adipocyte transdifferentiation [61]. Inflammatory cytokines such as TNF-α and IL-6 induce NF-κB activation in adipocytes, upregulating lipolytic enzymes (ATGL, HSL) and thermogenic markers such as UCP1 [62,63]. These changes drive elevated free fatty acid (FFA) release and energy expenditure, further aggravating the negative energy balance characteristic of cachexia [64]. IL-6 further amplifies these effects through the AMPK–HSL and JAK/STAT3 axes, while redox-sensitive mechanisms (e.g., ROS, TLRs) sustain NF-κB activation even in the absence of exogenous stimuli [65,66]. Therapeutically, NF-κB inhibition has shown promise in attenuating both muscle and fat loss [67]. Pharmacologic agents such as DHMEQ, SR12343, and NSAIDs have been reported to suppress inflammation, restore IGF-1/Akt/mTOR signaling, and reduce senescence-associated secretory phenotypes (SASP) in muscles [68]. In adipose tissue, NF-κB blockade via IKKβ inhibition or IκBα overexpression—reduces lipolysis, prevents browning, and preserves adipocyte integrity [69]. Targeting IL-6 signaling provides synergistic benefits by modulating upstream pathways [70,71]. These findings support a multifactorial therapeutic framework targeting the NF-κB centered inflammatory metabolic axis to restore tissue homeostasis, preserve energy stores, and delay cachexia progression [72]. Continued investigation into these strategies may yield novel combinatorial interventions with clinical translational potential [73].
4.2. NF-κB as a Central Mediator of Adipose Remodeling and Systemic Metabolic Dysregulation in Cancer Cachexia
Beyond its well-established role in muscle atrophy, NF-κB also orchestrates profound alterations in adipose tissue metabolism and systemic energy homeostasis during cancer cachexia [73]. In white adipose tissue (WAT), pro-inflammatory cytokines particularly TNF-α, IL-1β, and IL-6 activate the canonical IKKβ/NF-κB signaling pathway in adipocytes, leading to the upregulation of lipolytic enzymes such as adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) [74]. This accelerates triglyceride breakdown and promotes free fatty acid (FFA) release, thereby contributing to elevated energy expenditure and progressive fat loss [75]. Concurrently, NF-κB induces the expression of thermogenic genes like uncoupling protein 1 (UCP1), driving the browning of white adipose tissue and further enhancing futile energy dissipation [63,76]. IL-6 amplifies these effects by simultaneously engaging the AMPK–HSL axis and the JAK/STAT3 pathway, reinforcing both lipolysis and thermogenesis.
Therapeutically, both adipose- and muscle-specific NF-κB inhibition have shown efficacy in reversing cachexia-related metabolic disturbances in preclinical models [77]. Pharmacological agents such as NSAIDs (e.g., indomethacin) and direct NF-κB inhibitors (e.g., DHMEQ, SR12343) have been shown to restore glucose tolerance, improve insulin sensitivity, reduce lipolysis, and preserve adipocyte morphology [78,79,80]. However, given NF-κB’s role in tissue repair and immune defense, systemic inhibition must be carefully calibrated to avoid unwanted side effects, such as impaired regeneration or heightened infection risk [81]. Overall, these findings position NF-κB as a master integrator of inflammatory and metabolic networks in cancer cachexia, as well as a highly actionable target for restoring adipose function and systemic energy homeostasis [82].
4.3. NF-κB in Neuro-Metabolic Control of Cachexia
Anorexia is a cardinal feature of cancer cachexia that contributes to systemic energy imbalance and worsened clinical outcomes. Central to this process is the activation of hypothalamic NF-κB signaling, which integrates inflammatory and nutritional cues to regulate appetite. Circulating cytokines such as TNF-α, IL-6, and IL-1β cross the blood–brain barrier via active transport or leaky regions like the median eminence and activate NF-κB in hypothalamic neurons [83]. This activation suppresses orexigenic pathways (e.g., ghrelin–NPY/AgRP) and enhances anorexigenic signaling (e.g., POMC/α-MSH), leading to reduced food intake and persistent metabolic suppression. Experimental models have shown that lipopolysaccharide (LPS) or HIV-Tat protein activates NF-κB in POMC neurons, inducing anorexia, while targeted deletion of IKKβ in these neurons alleviates inflammation-induced appetite loss [83]. Beyond neurons, TLR4/NF-κB signaling in hypothalamic microglia and astrocytes drives neuroinflammation, impairs leptin and insulin sensitivity, and disrupts energy homeostasis [83,84]. Endoplasmic reticulum (ER) stress further engages the IKKβ/NF-κB pathway, linking nutrient overload to hypothalamic dysfunction. Given this multifaceted role, NF-κB presents a promising therapeutic target for reversing anorexia in cancer cachexia. Inhibition strategies ranging from natural compounds (e.g., curcumin, EPA) to pharmacologic agents (e.g., NSAIDs) and genetic interventions (e.g., astrocyte-specific IKKβ knockout) have been shown to reduce hypothalamic inflammation, restore orexigenic signaling, and improve food intake in preclinical models [85]. Notably, clinical studies demonstrate that curcumin alleviates anorexia-related symptoms and improves muscle strength in cachectic patients, while agents like CTRP4 restore leptin sensitivity via astrocytic NF-κB modulation [83,86]. Collectively, NF-κB orchestrates muscle catabolism, adipose remodeling, and neuro-metabolic dysregulation, converging on systemic energy imbalance and progressive cachexia (Figure 3).
5. NF-κB Inhibition as a Therapeutic Strategy in Cancer Cachexia
NF-κB is a central mediator of inflammation, proteolysis, and metabolic dysfunction in cancer cachexia, making it a highly promising therapeutic target. Several pharmacological agents including small molecule inhibitors, natural compounds, and anti-inflammatory drugs have demonstrated efficacy in preclinical and clinical studies. Key preclinical and clinical studies targeting NF-κB in cancer cachexia are summarized in Table 1. SR12343, a selective IKK/NF-κB inhibitor, significantly attenuated chemotherapy-induced muscle wasting by suppressing NF-κB activation, reducing senescence-associated secretory phenotype (SASP) markers, and preserving lean and fat mass, grip strength, and body weight [87]. Similarly, DHMEQ, which blocks NF-κB p65 DNA-binding activity, alleviated muscle atrophy and systemic inflammation in mouse models. Natural compounds such as curcumin and EPA also exhibit NF-κB inhibitory effects. In breast cancer cachexia models, curcumin downregulated MuRF1 and Atrogin-1, improved mitochondrial function, and preserved muscle fiber integrity [88]. NSAIDs, including indomethacin and ibuprofen, inhibit COX, thereby reducing prostaglandin synthesis and NF-κB-mediated inflammatory signaling. A systematic review indicated that prolonged NSAID use reduced the incidence of cancer cachexia by approximately 23% [89,90]. However, anti–TNF-α therapies (e.g., infliximab, etanercept) have shown inconsistent outcomes, likely due to cytokine redundancy and compensatory activation of IL-6 or IL-1β. In contrast, IL-6 receptor antagonists (e.g., tocilizumab, siltuximab) have demonstrated more consistent benefits, including suppression of systemic inflammation and improvement in muscle function through dual inhibition of NF-κB and STAT3 pathways [91,92,93].
Given the multifactorial nature of cachexia, combination therapies that concurrently target NF-κB and other pathogenic mechanisms are gaining prominence. Co-administration of SR12343 with chemotherapy reduced muscle atrophy, systemic inflammation, and SASP expression, highlighting its dual role in protecting muscle integrity and enhancing chemotherapy tolerance [59]. Similarly, the combination of DMAPT and cisplatin enhanced chemosensitivity and preserved body weight and renal function in bladder cancer models by reducing IL-6 elevation. Nutritional and anti-inflammatory regimens—such as combinations of niacin, celecoxib, L-carnitine, and EPA have improved lean mass, reduced CRP, TNF-α, and reactive oxygen species, and enhanced patient-reported outcomes [94]. Clinical trials have shown that ibuprofen supplementation can stabilize body mass in patients with lung cancer. Furthermore, dual inhibition of TGF-β and NF-κB has shown efficacy in reducing muscle loss and prolonging survival in pancreatic cancer cachexia models. Exercise, as a non-pharmacologic adjunct, promotes anti-inflammatory myokine expression, reverses systemic inflammation, and mitigates muscle wasting. Collectively, these findings support a multimodal therapeutic paradigm that integrates NF-κB inhibition with chemotherapy, anti-cytokine therapies, nutritional support, and behavioral interventions. This integrative approach holds strong potential to preserve skeletal muscle, restore metabolic homeostasis, and improve clinical outcomes in patients with cancer cachexia (Figure 4).
6. Challenges and Future Directions
Despite compelling evidence supporting NF-κB as a central driver of cancer cachexia, accumulating data indicate that its pathological role is highly context-dependent, varying across disease stages, tissue compartments, and cellular subtypes [17,95,96,97]. Here, we propose a unifying NF-κB centered cachectic cascade model, in which temporal and spatial features of NF-κB activation determine its pathological impact across organs. While transient NF-κB activation is essential for immune defense, tissue repair, and adaptive stress responses, sustained low-grade activation characteristic of chronic tumor-associated inflammation drives persistent catabolic signaling, metabolic dysregulation, and progressive tissue wasting [98]. This temporal dichotomy provides a mechanistic framework to reconcile the inconsistent clinical efficacy of systemic anti-inflammatory strategies, particularly anti-TNF-α therapies, despite robust preclinical benefits [4,99,100].
Importantly, NF-κB signaling exerts distinct and sometimes opposing effects depending on the cellular context. Within the proposed cachectic cascade, NF-κB functions as a multicellular signaling hub rather than a uniform effector. In skeletal muscle fibers, chronic activation of the canonical IKKβ–NF-κB pathway directly induces proteolytic gene expression (e.g., MuRF1, Atrogin-1, iNOS) and suppresses anabolic signaling, thereby promoting muscle atrophy [3,13,101]. In contrast, NF-κB activation within infiltrating macrophages, fibro-adipogenic progenitors, and endothelial cells amplifies paracrine cytokine release and extracellular matrix remodeling, indirectly exacerbating muscle degeneration, supporting a multicellular regulation of wasting states [102]. Similarly, hypothalamic NF-κB activation integrates peripheral inflammatory signals to suppress appetite and disrupt neuroendocrine control of energy homeostasis, further accelerating systemic wasting [103]. Together, these observations highlight a spatiotemporally coordinated, multicellular inflammatory network in which NF-κB integrates local and systemic signals to drive cachexia progression [104].
From a translational perspective, this complexity highlights the limitations of indiscriminate NF-κB inhibition. Broad systemic blockade fails to account for disease stage–specific and tissue-specific NF-κB functions and may compromise immune surveillance and regenerative capacity, particularly in patients receiving cytotoxic chemotherapy [105,106]. Consequently, future therapeutic strategies should prioritize spatiotemporal precision, targeting pathological NF-κB activation within specific tissues or disease stages while preserving its physiological functions [107]. Such precision-based approaches represent a conceptual shift from pathway suppression toward context-aware modulation of inflammatory signaling. Approaches such as cell-type-restricted IKKβ modulation, combination therapies targeting upstream cytokine drivers (e.g., IL-6/JAK/STAT3), or integration with metabolic and exercise-based interventions may provide superior efficacy with reduced toxicity [108,109]. Consistent with this notion, the multifactorial nature of cancer cachexia has increasingly shifted therapeutic efforts toward multimodal strategies that simultaneously target inflammatory, metabolic, and oncogenic pathways [110]. Preclinical studies strongly support this paradigm. For instance, co-treatment with the NF-κB inhibitor DMAPT and cisplatin enhanced tumor chemosensitivity while attenuating chemotherapy-induced muscle wasting and systemic IL-6 elevation, preserving both body weight and renal function [111]. Similarly, the IKK/NF-κB inhibitor SR12343, when combined with chemotherapy, significantly reduced muscle atrophy, weight loss, and inflammation by suppressing senescence-associated secretory phenotype (SASP) signaling, highlighting the therapeutic relevance of targeting inflammation-induced senescence under cytotoxic stress [59]. Early-phase clinical interventions have mirrored these findings: regimens combining NSAIDs (e.g., ibuprofen, celecoxib) with metabolic supplements have demonstrated improvements in lean mass and reductions in systemic inflammation [99,112]. Emerging strategies further emphasize synergistic pathway modulation. Dual inhibition of TGF-β and NF-κB signaling in pancreatic cancer cachexia, for example, has demonstrated efficacy in preserving skeletal muscle, mitigating weight loss, and prolonging survival in preclinical models [113,114]. Collectively, these findings reinforce the necessity of addressing tumor burden, chronic inflammation, and metabolic dysregulation in parallel. Moving forward, successful clinical translation will depend on the precise optimization of treatment timing, dosage, and patient stratification to maximize therapeutic benefit while minimizing adverse effects particularly those that may compromise anabolic or regenerative pathways.
Finally, patient stratification based on inflammatory burden may be critical for the success of NF-κB targeted interventions. We propose a clinically actionable stratification framework in which biomarkers such as circulating IL-6, C-reactive protein, or transcriptional NF-κB activity signatures are used to identify patient subsets most likely to benefit from NF-κB modulation. As mechanistic understanding of cachexia deepens, future therapeutic regimens will likely adopt a precision medicine framework, integrating targeted molecular therapies with individualized nutritional support and lifestyle interventions. This approach reframes NF-κB targeted therapy from a palliative measure to a proactive, mechanism-based intervention strategy for cancer cachexia.
7. Conclusions
NF-κB functions as a central inflammatory metabolic regulatory hub in cancer cachexia, linking chronic systemic inflammation to skeletal muscle atrophy, impaired regenerative capacity, and global energy imbalance [54]. Pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 activate NF-κB-dependent signaling programs that drive transcription of catabolic genes, including MuRF1, Atrogin-1, and iNOS, thereby promoting activation of the ubiquitin proteasome system and autophagy lysosome pathway to accelerate muscle protein degradation and fiber atrophy [22,115]. In experimental models of cancer cachexia, sustained activation of the canonical NF-κB pathway disrupts muscle homeostasis not only by enhancing proteolysis but also by impairing regeneration, through increased Pax7 expression and inhibition of MyoD driven myogenesis [9]. Beyond skeletal muscle, NF-κB contributes to systemic metabolic dysfunction by repressing the IGF-1/Akt/mTOR anabolic pathway, promoting hepatic gluconeogenesis, inducing insulin resistance, and enhancing lipolytic enzyme activity [79,116]. It also facilitates white adipose tissue browning through UCP1 upregulation, further increasing energy expenditure [117]. These events reinforce a feed-forward inflammatory loop, in which NF-κB-driven catabolic and metabolic alterations amplify cytokine production, sustaining chronic low-grade inflammation. Collectively, current evidence derived predominantly from mechanistic and preclinical studies supports a prominent role for NF-κB as a coordinating signaling axis in cancer cachexia, while underscoring the need for further clinical validation to define its causal contribution and therapeutic tractability. Growing evidence supports inhibition of NF-κB related inflammatory signaling as a potentially viable therapeutic strategy for cancer cachexia, particularly when integrated with chemotherapy, anti-inflammatory agents, or metabolic support [118]. The IKK/NF-κB inhibitor SR12343 significantly attenuated chemotherapy-induced muscle wasting, preserved lean mass, and reduced SASP markers in murine models, while similar protective effects were observed with DMAPT cisplatin co-treatment [119]. Importantly, these benefits have been demonstrated predominantly in preclinical settings. In contrast, clinical regimens incorporating nonsteroidal anti-inflammatory drugs (NSAIDs), omega-3 polyunsaturated fatty acids, and metabolic supplements have shown more modest but reproducible benefits, including stabilization of body weight, reduction in systemic inflammatory markers, and improvement in quality-of-life measures in patients with advanced malignancies [120]. Furthermore, targeting convergent inflammatory nodes such as combined inhibition of NF-κB and TGF-β signaling has shown additive benefits in pancreatic cancer cachexia models, including improved survival [114]. Collectively, these findings support integrated, multimodal treatment paradigms targeting NF-κB-centered inflammatory–metabolic networks, while also underscoring the need for rigorously designed, large-scale randomized clinical trials to define optimal timing, dosing, safety profiles, and patient stratification strategies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Blum D. Stene G.B. Solheim T.S. Fayers P. Hjermstad M.J. Baracos V.E. Fearon K. Strasser F. Kaasa S. Validation of the Consensus-Definition for Cancer Cachexia and evaluation of a classification model—A study based on data from an international multicentre project (EPCRC-CSA)Ann. Oncol.2014251635164210.1093/annonc/mdu 08624562443 · doi ↗ · pubmed ↗
- 2Yeom E. Yu K. Understanding the molecular basis of anorexia and tissue wasting in cancer cachexia Exp. Mol. Med.20225442643210.1038/s 12276-022-00752-w 35388147 PMC 9076846 · doi ↗ · pubmed ↗
- 3Cai D. Frantz J.D. Tawa N.E.Jr. Melendez P.A. Oh B.C. Lidov H.G. Hasselgren P.O. Frontera W.R. Lee J. Glass D.J. IK Kbeta/NF-kappa B activation causes severe muscle wasting in mice Cell 200411928529810.1016/j.cell.2004.09.02715479644 · doi ↗ · pubmed ↗
- 4Webster J.M. Kempen L. Hardy R.S. Langen R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia Front. Physiol.20201159767510.3389/fphys.2020.59767533329046 PMC 7710765 · doi ↗ · pubmed ↗
- 5Langen R.C. Haegens A. Vernooy J.H. Wouters E.F. de Winther M.P. Carlsen H. Steele C. Shoelson S.E. Schols A.M. NF-κB activation is required for the transition of pulmonary inflammation to muscle atrophy Am. J. Respir. Cell Mol. Biol.20124728829710.1165/rcmb.2011-0119 OC 22538866 PMC 5460909 · doi ↗ · pubmed ↗
- 6Knaevelsrud H. Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins FEBS Lett.20105842635264510.1016/j.febslet.2010.04.04120412801 · doi ↗ · pubmed ↗
- 7Yadav A. Singh A. Phogat J. Dahuja A. Dabur R. Magnoflorine prevent the skeletal muscle atrophy via Akt/m TOR/Fox O signal pathway and increase slow-My HC production in streptozotocin-induced diabetic rats J. Ethnopharmacol.202126711351010.1016/j.jep.2020.11351033141056 · doi ↗ · pubmed ↗
- 8Zimmers T.A. Fishel M.L. Bonetto A. STAT 3 in the systemic inflammation of cancer cachexia Semin. Cell Dev. Biol.201654284110.1016/j.semcdb.2016.02.00926860754 PMC 4867234 · doi ↗ · pubmed ↗