An Antioxidant Cocktail of tert-Butylhydroquinone and a Manganese Porphyrin Induces Toxic Levels of Oxidative Stress in Cancer Cells
Sandra Tamarin, Hannah Jung, Joseph LaMorte, Laura Biesterveld, Gabriel Piñero, Grace Turchetta, Molly S. Myers, Rebecca Oberley-Deegan, Aimee L. Eggler

TL;DR
A combination of two antioxidants causes cancer cell death by creating toxic oxidative stress, while leaving healthy cells unharmed.
Contribution
A novel antioxidant cocktail that selectively induces oxidative stress and apoptosis in cancer cells.
Findings
The combination of tBHQ and MnBuOE increases oxidative stress and causes apoptosis in cancer cells.
The electrophilic quinone tBQ is critical for toxicity, while non-electrophilic dtBQ is non-toxic.
Hydrogen peroxide generation is essential for the observed cell death.
Abstract
Despite significant advancement in cancer treatments, therapies with minimal toxicity to healthy cells are still limited. One targetable weakness of cancer cells is their sensitivity to oxidative stress. We find that the combination of two antioxidants—the common food additive tert-butylhydroquinone (tBHQ) and a manganese porphyrin in clinical trials, MnTnBuOE-2-PyP5+ (MnBuOE)—increases oxidative stress and causes apoptotic death in several cancer cell lines, but not in mouse primary fibroblasts. Investigating the mechanism of cell death, MnBuOE is observed to catalyze the oxidation of tBHQ, producing the electrophilic quinone tert-butylquinone (tBQ). A critical role for tBQ and its electrophilic character was revealed with the observation that di-tert-butylhydroquinone (dtBHQ) in combination with MnBuOE causes no observable oxidative stress and is non-toxic, despite rapid oxidation to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
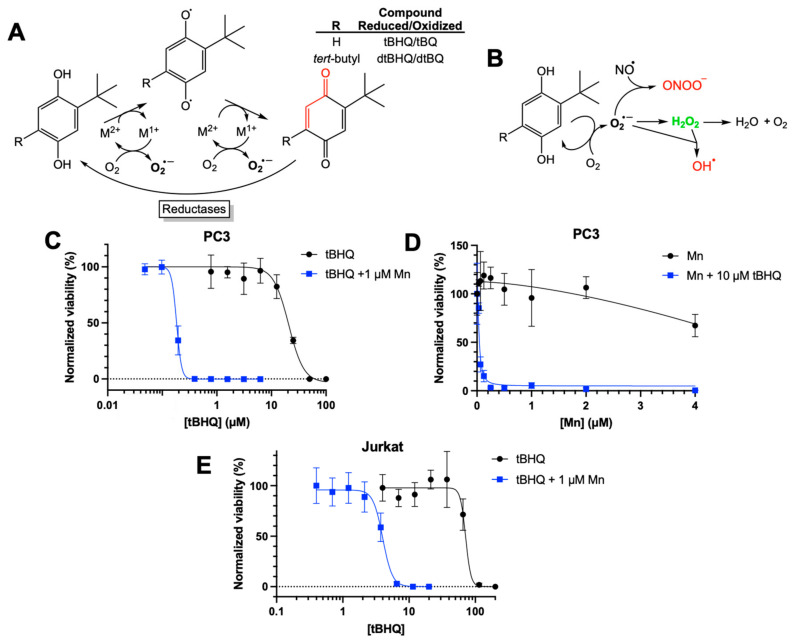 Figure 1
Figure 1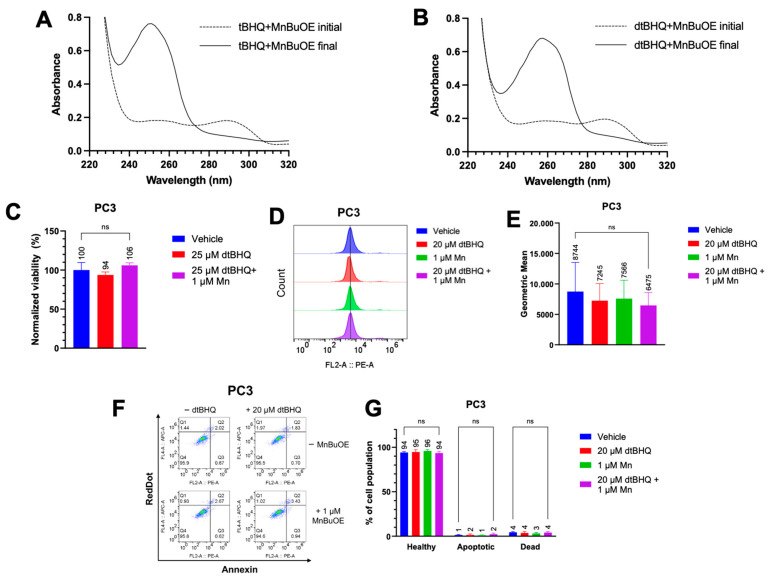 Figure 4
Figure 4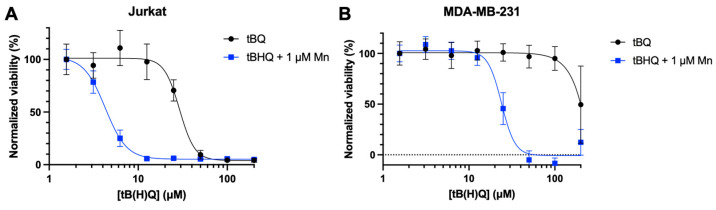 Figure 5
Figure 5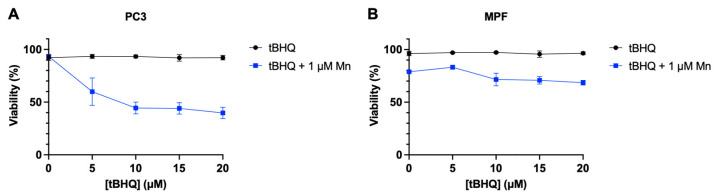 Figure 6
Figure 6- —National Institutes of Health
- —Villanova University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBioactive Compounds and Antitumor Agents · Free Radicals and Antioxidants · Photodynamic Therapy Research Studies
1. Introduction
Cancer remains a common cause of death worldwide, with a projected global economic cost of USD 25.2 trillion in international dollars between the years 2020 and 2050 [1]. Traditional treatments, such as chemotherapy, can cause debilitating side effects [2]. Newer targeted and personalized treatments such as monoclonal antibodies [3] and cell therapies [4,5] are having some success but are cost-prohibitive [6,7], and their effectiveness is limited to tumors that express certain target molecules [3]. Therapies that are inexpensive, effective, and minimize off-target effects are crucial for the future of cancer treatment. One strategy to target cancer cells takes advantage of their weakness to induced oxidative stress at levels that are non-toxic to healthy tissues. For example, cancer cells are susceptible to the oxidative stress generated by ascorbic acid, a well-known antioxidant that can also produce large amounts of reactive oxygen species (ROS) in the presence of catalytic metal ions [8].
We observed a toxic effect on cancer cell lines from two small molecules in combination: tert-butyl hydroquinone (tBHQ) and a manganese porphyrin, each best known for their antioxidant effect. tBHQ is a phenolic compound that is widely used as a food additive to mitigate fat oxidation [9]. It is considered safe for use in this capacity by the FDA and the WHO [10,11,12]. While studies have demonstrated that tBHQ can have toxic effects in cancer cells, inducing apoptosis and causing DNA damage [13,14], toxic concentrations are generally considered well above the physiological range attainable by oral dosing. For example, studies in animals show that while 80–90% of orally dosed tBHQ is bioavailable, it is rapidly metabolized in the liver to the 4-O-sulphate conjugate and the 4-O-glucuronide conjugate [9,15,16], greatly minimizing free tBHQ.
First developed as superoxide dismutase mimics, manganese porphyrins have been optimized for over three decades for bioavailability, minimized toxicity, and therapeutic efficacy, and myriad interactions with redox-sensitive pathways have been identified [17]. They protect normal cells from the harmful effects of radiation therapy, while at the same time, sensitize cancer cells to the therapy [18,19]. They also tend to accumulate at higher concentrations in cancer cells, as opposed to healthy cells [19], and these traits have made them promising therapeutic agents. The manganese porphyrin tested in this study, MnTnBuOE-2-PyP^5+^ (MnBuOE), also called BMX-001, is currently being assessed in clinical trials as a radioprotectant of normal tissues during treatment for cancers ranging from head and neck to rectal. Its effectiveness is evident in a recently completed Phase II glioma trial, in which BMX-001 was tested in combination with the radiation and oral chemotherapy drug temozolomide, where the data showed that BMX-001 improved overall patient survival [19,20].
Herein, we present the effect of the combination of tBHQ and MnBuOE on cancer cells. We show that, while each antioxidant on its own is toxic only at relatively high doses, the combination of both drugs is toxic to cancer cells at physiologically achievable doses. We investigate the mechanism by which this cell death occurs and propose that the toxicity is caused by hydrogen peroxide generated by tert-butylquinone (tBQ), the electrophilic quinone produced by the manganese porphyrin-catalyzed oxidation of tBHQ. Additionally, we demonstrate the ability of this combination treatment to target cancer cells by showing its minimal toxicity in non-cancerous primary cells.
2. Materials and Methods
2.1. Chemicals
Unless stated otherwise, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). The compounds tBHQ (112941), tBQ (429074), and di-tert-butylhydroquinone (dtBHQ) (112976) were prepared daily in DMSO (472301) from powder to 50 mM. Both tBHQ and dtBHQ were prepared in fresh plastic tubes, and the ultrapurified water used in subsequent dilutions was stored in the same, to prevent trace metal contamination. MnBuOE, a gift from Dr. James Crapo, was dissolved in PBS pH 7.4 (consisting of 137 mM NaCl (S9888), 2.7 mM KCl (P4504), 10 mM Na_2_HPO_4_ (J.T. Baker, 302801, Phillipsburg, NJ, USA), and 1.8 mM KH_2_PO_4_ (VWR, BDH0268, Radnor, PA, USA) to 4 mM, with aliquots stored in the dark at 4 °C. Manganese (III) tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP) (Santa Cruz Biotechnology, SC-221956, Dallas, TX, USA) was prepared in the same manner. Treatments contained 1 µM MnBuOE unless otherwise stated. Catalase (C1345) was prepared daily by dissolving in 50 mM phosphate followed by sterile filtration, and the final concentration was 2000 U/mL in treated wells.
2.2. UV–Vis Spectroscopy
Solutions of 25 µM tBHQ or dtBHQ in PBS at pH 7.4 were prepared, and their UV–Vis spectra from 220 to 520 nm were measured on an Agilent Cary 60 UV–Vis spectrophotometer (Santa Clara, CA, USA) at room temperature (21 °C). To observe oxidation of each to the quinone form, 30 µL of 0.1 mM MnBuOE was added to 3 mL final volume of the tBHQ or dtBHQ solutions, and spectra were collected every 30 to 60 s. To maintain solubility of the di-tert-butylquinone (dtBQ) product at 21 °C upon its formation, reactions were set up under the same conditions but with 200 proof ethanol included at 50% final concentration. Data were fit to a one phase decay using GraphPad Prism version 10.6.1 (San Diego, CA, USA).
2.3. Cell Culture
2.3.1. Suspension Cell Culture
The Jurkat clone E6-1 cell line (TIB-152, ATCC, Manassas, VA, USA) was maintained at 37 °C in 5% CO_2_ in RPMI-1640 containing phenol red (Cytiva, SH30096.01, Marlborough, MA, USA) and stabilized L-glutamine (L-alanyl-L-glutamine dipeptide) (bioWORLD, 44043001-1, Dublin, OH, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (R&D Bio-Techne, S1115014, Minneapolis, MN, USA) in T75 flasks. This complete media was stored at 4 °C, with aliquots warmed to 37 °C just prior to use. Cell cultures were passaged every two to four days and were strictly maintained at cell densities between 1 × 10^5^ and 1.5 × 10^6^ cells/mL during propagation.
2.3.2. Adherent Cell Culture
The PC3 cell line (CRL-1435, ATCC) and the MDA-MB-231 (HTB-26, ATCC) cell line were grown in complete media in T75 flasks. Media for these lines was RPMI and Dulbecco’s Modified Eagle Medium (DMEM) (GenClone, 25-501N, El Cajon, CA, USA), respectively, both supplemented with 10% FBS. For experiments comparing PC3 cells to mouse prostate fibroblast (MPF) cells, 1% penicillin/streptomycin (Cytiva, SV30010) was also included in the cell culture media. Cells were passaged when they had reached 70–85% confluency.
2.3.3. Mouse Primary Prostate Fibroblast Isolation and Cell Culture
Male C57BL/6J mice obtained from Jackson Laboratories (Bar Harbor, ME, USA) were used in this study [21]. The mice were housed at the University of Nebraska Medical Center (UNMC) and exposed to a 12 h light/12 h dark cycle and fed and watered ad libitum. This study was carried out in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All procedures were approved by the institutional animal care and use committee at UNMC (20-019-03-FC).
Mouse primary fibroblasts were isolated as previously described [21]. Briefly, prostates were isolated from 6 to 8 week old C57BL/6J mice. Tissue was minced with a scalpel then digested in 5 mg/mL collagenase I (Thermo Fisher, 17100017, Waltham, MA, USA) for 30 min at 37 °C. Cells and tissue fragments were then cultured for 2–3 weeks in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% nonessential amino acids (Thermo Fisher, 11140050). Cultures were maintained at 5% CO_2_ and 37 °C. After 5 days of culture, only fibroblasts were present.
2.4. Toxicity Assays
2.4.1. Cell Treatments
Cells were treated for 24 h with tBHQ, dtBHQ, dtBQ, MnBuOE, and/or catalase, as indicated in the figures. The final volume in all wells was 105 µL, and the final DMSO concentration in media was 0.05%. When designing plate layouts for experiments, care was taken to minimize any neighboring well effect of the treatments, as previously reported for tBHQ [22]. Thus, titrations of tBHQ, dtBHQ, or tBQ were set up from lowest to highest concentration across a plate. The term “technical replicates” in corresponding figure legends refers to multiple wells treated individually on the same day from the same passage number cells.
2.4.2. CellTiter-Glo® Assay
The toxicities of tBHQ, dtBHQ, and MnBuOE in PC3 and Jurkat cells were determined using a CellTiter-Glo^®^ assay, which measures ATP levels and defines a living cell as a metabolically active one. After treatment, cells were washed with PBS, followed by addition of 25 µL of PBS and 25 µL of CellTiter-Glo 2.0 (CTGlo) (Promega, G9241, Madison, WI, USA), and incubation in the dark at room temperature for 10 min. Luminescence was measured using a ClarioStar Plus plate reader (BMG Labtech, Ortenberg, Germany).
2.4.3. CellTiter-Fluor™ Assay
Toxicities of tBHQ, tBQ, and MnBuOE in Jurkat cells were determined using a CellTiter-Fluor™ assay, which assesses viable cells by measuring protease activity. After treatment, cells were washed with PBS, followed by resuspension in 25 µL Hank’s Buffered Salt Solution (HBSS) (Cytiva, SH30268.01). Upon addition of 25 µL of CellTiter-Fluor (CTF) (Promega, G6080), samples were incubated in the dark at 37 °C for 1 h. Fluorescence was measured using the ClarioStar Plus plate reader with excitation and emission of 390/505 nm.
2.4.4. Bicinchoninic Acid (BCA) Assay
Toxicities of tBHQ, tBQ, and MnBuOE in MDA-MB-231 cells were determined using a BCA assay, which, after washing adhered cells to remove lysed cells and their concentration, measures remaining living cells by total protein content. After treatment, cells were washed twice with PBS, followed by addition of 25 µL lysis buffer consisting of 0.03% Triton X-100 (X100RS). After incubation on a plate shaker for 10 min, 200 µL of BCA reagent (Thermo Fisher, 23225) was added, and the plate was shaken for 3 min. The plate was incubated at 37 °C for 30 min. Absorbance was measured at 562 nm on the ClarioStar Plus plate reader.
2.4.5. Data Analysis to Determine the Concentration Lethal to 50% of the Cells (LC50)
Readings from each assay were either normalized to the untreated control or to the lowest concentration of the cells treated with tBHQ, as described in the figure legends. Data were analyzed using GraphPad Prism, and the LC50 values were determined using a variable-slope four-parameter fit. The data were analyzed without constraining the parameters, except for tBHQ + MnBuOE in Figure 1C (top constrained to 100) and the data in Figure 1E (bottom constrained to 0).
2.4.6. Trypan Blue Exclusion Assay
A Trypan blue exclusion assay was used to compared the toxicity of tBHQ and MnBuOE in PC3 versus MPF cells. After treatment, cells were trypsinized and pelleted, then resuspended in 200 µM of cell culture media. Cells were counted and viability was measured using a Vi-CELL BLU cell viability analyzer (Beckman Coulter, Brea, CA, USA).
2.5. Flow Cytometry Assays
2.5.1. Cell Treatment
PC3 cells in a 24-well plate at ~80% confluency were treated in 500 µL of media with 20 µM tBHQ or dtBHQ and with 1 µM MnBuOE where indicated. Jurkat cells (1 × 10^6^ cells/mL in 150 µL) in a 96-well plate were treated with 10 µM tBHQ or dtBHQ and with 1 µM MnBuOE where indicated. The term “technical replicates” in corresponding figure legends refers to multiple wells treated individually on the same day from the same passage number cells. The term “biological replicates” refers to experiments conducted on different days with a different passage number.
After 4 h in a 37 °C CO_2_ incubator, PC3 cells were trypsinized to remove them from the plate, and Jurkat cells were removed directly from the plate for the apoptosis and necrosis assay. Following completion of this assay, after 6 h total of treatment time, additional wells were used to harvest PC3 cells and Jurkat cells for the MitoSOX assay.
2.5.2. Apoptosis and Necrosis Assay
Apoptosis and necrosis were assayed by flow cytometry using Annexin-PE (Biotium, 29045, Fremont, CA, USA) to measure apoptosis and RedDot 2 dye (Biotium, 40061-T) to measure necrosis, according to manufacturer instructions. A sample (nominally comprising 1 × 10^5^ cells) was centrifuged and washed with 100 µL of Annexin binding buffer (10 mM HEPES (GoldBio, H-400, St. Louis, MO, USA), 140 mM NaCl, and 2.5 mM CaCl_2_ (Acros Organics, 423525000, Geel, Belgium), pH 7.4). Cells were resuspended in 20 µL of this buffer with 1 µL of Annexin and 0.05 µL of RedDot added and incubated at room temperature in the dark for 10 min. An additional 80 µL of Annexin binding buffer was added, and the cells were incubated for an additional 10 min covered from the light. Fluorescence was measured using an Accuri C6 Plus flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). A 488 nm laser was used to excite the dyes, and emitted fluorescence was measured using the FL2 (585/40 nm) and FL4 (675/25 nm) filters for Annexin and RedDot, respectively. A total of 10,000 events were collected for each sample. The data were analyzed using FlowJo version 10.10.0 (Ashland, OR, USA).
2.5.3. MitoSOX Red Assay
MitoSOX Red, which is targeted to the mitochondria and fluoresces when oxidized, was used according to manufacturer instructions. A sample (nominally comprising 1 × 10^5^ cells) was centrifuged and washed twice with 100 µL of Annexin binding buffer. Cells were then resuspended in 100 µL Annexin binding buffer containing 1 µM MitoSOX Red (Invitrogen, M36008, Waltham, MA, USA). The cells were covered from the light and incubated for 10 min at room temperature, then centrifuged and resuspended in 100 µL binding buffer. The cells were incubated for another 10 min covered from light, and then fluorescence was measured using an Accuri C6 Plus flow cytometer. A 488 nm laser was used to excite the dyes, and emitted fluorescence was measured using the FL2 filter. Unless otherwise noted, a total of 10,000 events were collected for each sample. The data were analyzed using FlowJo version 10.10.0.
2.6. Statistical Analysis
Statistical analysis was performed using GraphPad Prism. For the MitoSOX experiments and the CTGlo experiments that compared single concentrations of various additives and were presented in bar graph format, a one-way ANOVA test was used, followed by Dunnett’s multiple comparisons test to compare each condition to the vehicle control. For the apoptosis/necrosis flow cytometry results, a two-way ANOVA test was used, followed by Dunnett’s multiple comparisons test to compare each condition to the vehicle control. A p value of less than 0.05 was considered significant.
3. Results
3.1. The Combination of tBHQ and MnBuOE Is More Toxic than Either Component Alone
While previously investigating the toxicity of tBHQ as a redox-cycling compound (Figure 1A), we attempted to mitigate this toxicity to immortalized cell lines by including a commercially available superoxide dismutase mimetic—MnTMPyP. As shown in Figure 1A, hydroquinones such as tBHQ and dtBHQ are readily oxidized in a catalytic manner by transition metals to first the semiquinone and then the quinone, generating superoxide. Toxicity from redox cycling is generally attributed to reactive oxygen and nitrogen species formed downstream of superoxide (Figure 1B). Superoxide dismutase and its small molecule mimetics act as antioxidants by catalyzing the conversion of superoxide to hydrogen peroxide, limiting generation of strong oxidizing molecules such as the hydroxyl radical and peroxynitrite. Previous studies showed that MnTMPyP was highly effective at rescuing dtBHQ-induced suppression of global protein synthesis in the immortalized HaCaT keratinocyte cell line [23], and we expected the same for tBHQ-induced effects when treating with MnTMPyP. Instead, we observed that MnTMPyP greatly sensitized HaCaT cells and the adherent breast cancer cell line MDA-MB-231 to tBHQ, significantly decreasing tBHQ LC50 values (Figure S1).
To determine whether this observation might be translatable to a therapeutically relevant manganese porphyrin in clinical trials, the combination of MnBuOE and tBHQ was investigated in cancer cell lines. As shown in Figure 1C, in PC3 cells, an adherent prostate cancer cell line, tBHQ had an LC50 value of 21 ± 1 µM. MnBuOE alone also showed low toxicity at concentrations of 2 µM or less (Figure 1D). However, the combination of tBHQ with 1 µM MnBuOE resulted in an LC50 of 0.2 ± 0.02 µM, a 100-fold increase in the potency of tBHQ. Experiments performed with a different passage number showed very similar results (Figure S2A). Thus, the combination of tBHQ and MnBuOE was far more toxic to PC3 cells than either component by itself. A titration of MnBuOE with 10 µM tBHQ showed that 0.25 µM MnBuOE was sufficient for the toxic effect (Figure 1D), which is far less than the amount of MnBuOE (2–6 µM) that has been shown to accumulate in the liver, kidney, and lymph nodes of mice [24].
To discover if this phenomenon applied to other cancer cell lines, we tested the combination in Jurkat cells, a suspension leukemia cell line. As with PC3 cells, tBHQ alone was fairly non-toxic to Jurkat cells, with an LC50 value of 72 ± 10 µM (Figure 1E). Inclusion of MnBuOE decreased the LC50 approximately 18-fold to 4 ± 0.2 µM. Additionally, testing the combination treatment in MDA-MB-231 cells showed similar results, reducing the LC50 by approximately 10-fold (Figure S3).
Toxic effects of combining tBHQ and a manganese porphyrin superoxide dismutase mimetic. (A) The two-electron redox cycling of tBHQ and dtBHQ, catalyzed by transition metals, produces superoxide. Oxidation by one electron produces the semiquinone, with loss of the second electron producing the quinone. While the tBQ quinone is an electrophilic α,β unsaturated carbonyl (indicated in red), the corresponding carbon in dtBQ is sterically blocked from reacting with a nucleophile by the bulky t-butyl group. Cellular reductases can reduce a quinone by two electrons back to the hydroquinone, allowing for redox cycling with superoxide as a product. (B) Upon production of superoxide in redox cycling, subsequent reactions and transformations result in biologically relevant reactive oxygen and nitrogen species. In particular, the highly spontaneous reaction of nitric oxide with superoxide creates the damaging oxidant peroxynitrite, and the rapid conversion of superoxide to hydrogen peroxide catalyzed by superoxide dismutase is critical to preventing this. The metal-catalyzed Haber–Weiss reaction of superoxide with hydrogen peroxide generates the most damaging of all reactive oxygen species, the hydroxyl radical. A superoxide dismutase mimetic would be expected to significantly limit production of peroxynitrite and hydroxyl radical by more fully converting superoxide to hydrogen peroxide. (C) PC3 cells were treated with increasing concentrations of tBHQ alone or in combination with MnBuOE. Viability was measured using a CellTiter Glo assay and normalized to the untreated control. (D) PC3 cells were treated with increasing concentrations of MnBuOE alone or in combination with 10 µM tBHQ. Viability was measured using a CellTiter Glo assay and normalized to the untreated control. (E) Jurkat cells were treated with increasing concentrations of tBHQ alone or in combination with 1 µM MnBuOE. Viability was measured using a CellTiter Glo assay and normalized to the untreated control. n = 5 (technical replicates). “Mn” in the legends for (C–E) is the abbreviation for “MnBuOE”.
3.2. Hydrogen Peroxide Is a Major Contributor to the Toxicity of the tBHQ–MnBuOE Treatment
We hypothesized that the toxicity of the tBHQ–MnBuOE combination was caused by oxidation of tBHQ catalyzed by the manganese ion in MnBuOE (Figure 1A). Therefore, we examined the extent to which the tBHQ–MnBuOE combination induced oxidative stress in PC3 and Jurkat cells. Because MnBuOE can localize to the mitochondria [19], MitoSOX Red dye was used as a measure of oxidative stress in the mitochondria, noting that dye uptake and thus signal is dependent on mitochondrial membrane potential and mitochondrial mass. Thus, a caveat of the assay is that a treatment that reduces mitochondrial membrane potential and/or mass could result in lower dye uptake and lower signal. However, in PC3 cells, neither 20 µM tBHQ nor 1 µM MnBuOE alone changed the signal intensity of MitoSox Red dye in the cells, indicating that neither compound induces oxidative stress in cells compared to vehicle (Figure 2A,B). Moreover, the combination of tBHQ and MnBuOE increased the MitoSOX signal intensity by approximately four-fold compared to the vehicle, shown by an increase in the geometric mean (Figure 2A,B). Jurkat cells showed similar results, with an approximately three-fold signal increase in cells treated with tBHQ and MnBuOE compared to vehicle (Figure 2C,D). Thus, in both cell types, increased signal in response to the combination treatment indicated that oxidative stress increased.
As hydrogen peroxide is generally considered to be a major contributor to cell stress caused by two-electron redox cycling (Figure 1A), catalase was included at the time of treatment of PC3 cells with 10 µM tBHQ and MnBuOE. Catalase converts hydrogen peroxide into harmless water and oxygen. Although catalase does not enter the cell, it reduces intracellular hydrogen peroxide levels due to the ability of hydrogen peroxide to cross cellular membranes [25,26]. Addition of catalase improved cell viability from ~25% to ~90% in cells treated with the tBHQ–MnBuOE combination (Figure 2E). Experiments performed with cells at a different passage number showed the toxicity to PC3 cells caused by the combination of 25 µM tBHQ and 1 µM MnBuOE was prevented by the inclusion of catalase (Figure S2B). These data suggest that hydrogen peroxide is a major factor in the mechanism of cell death caused by the tBHQ–MnBuOE combination.
3.3. The Combination of tBHQ and MnBuOE Causes Apoptotic Cell Death
Since oxidative stress has been linked to apoptotic cell death [27], early apoptosis was measured in PC3 and Jurkat cells treated with tBHQ and MnBuOE using an Annexin V/RedDot assay. As expected, each component alone did not cause an increase in apoptosis or necrosis (Figure 3A,B). However, PC3 cells treated with a combination of 20 µM tBHQ and 1 µM MnBuOE showed an increase in apoptotic cell death of approximately 15% compared to cells treated only with vehicle. Necrotic cell death also increased to approximately 20%, indicating that there is some other mechanism of cell death occurring as well. Jurkat cells treated with 10 µM tBHQ and 1 µM MnBuOE also showed an increase in apoptotic cell death of nearly 20%, but no significant increase in necrotic cell death (Figure 3C,D), indicating that oxidative stress was the most likely cause of cell death.
3.4. Cell Death Is Related to the Catalyzed Oxidation of tBHQ into an Electrophilic Quinone
The data strongly suggest that hydrogen peroxide is a major contributor to cell death caused by the tBHQ–MnBuOE treatment. To investigate the extent to which hydroquinone redox cycling or perhaps the electrophilic nature of tBQ were responsible for generating hydrogen peroxide at toxic levels, dtBHQ was tested in combination with MnBuOE. dtBHQ is a useful analog of tBHQ, with an additional tert-butyl group (Figure 1A) that blocks the addition of nucleophiles to the quinone produced upon oxidation, dtBQ [28,29]. dtBHQ is readily oxidized to dtBQ in the presence of transition metals.
We first determined whether MnBuOE was able to oxidize both tBHQ and dtBHQ to their quinone forms, using UV–Vis spectroscopy. Both tBHQ and dtBHQ at 25 µM readily oxidized upon addition of MnBuOE (Figure S6A,B). However, as dtBQ accumulated, its lower water solubility than tBQ, together with a room temperature of 21 °C, caused precipitation, shown by an increase in light scattering at 500 nm (Figure S6B), complicating comparison of tBHQ and dtBHQ oxidation rates. Conducting both reactions in 50% ethanol to solubilize the dtBQ at room temperature, the kinetics of oxidation for tBHQ and dtBHQ were assessed, with similar kobs for each (the oxidation of tBHQ is two times faster than that of dtBHQ) (Figure S6C–F). The first and last timepoint spectra for tBHQ and dtBHQ with MnBuOE are shown in Figure 4A and Figure 4B, respectively. These results suggest that MnBuOE will readily catalyze the oxidation of both compounds in the cellular environment, and thus that hydrogen peroxide from redox cycling will be generated when either tBHQ or dtBHQ is present in combination with MnBuOE.
To test whether redox cycling-generated hydrogen peroxide was responsible for the toxicity to cancer cells, PC3 cells were treated with MnBuOE and dtBHQ at a concentration of dtBHQ (25 µM) that was highly toxic in parallel experiments with tBHQ in the combination treatment (Figure 1C). Either with dtBHQ alone or in combination with MnBuOE, there was no statistically significant decrease in cell viability (Figure 4C). Similar results were observed in Jurkat cells treated with 15 µM dtBHQ alone, 2 µM MnBuOE alone, or their combination (Figure S4E). Likewise, treating PC3 cells with 20 µM dtBHQ and 1 µM MnBuOE alone or in combination resulted in no increase in mitochondrial oxidative stress, apoptosis, or necrosis (Figure 4D–G). Similar results were seen in Jurkat cells treated with 10 µM dtBHQ and 1 µM MnBuOE (Figure S5). Therefore, two-electron redox cycling by dtBHQ likely generates insufficient hydrogen peroxide to induce cell death. By extension, redox cycling by tBHQ is likely insufficient to cause cell death. Instead, as for other arylating quinones [30], the cell death that occurs when cells are treated with tBHQ and MnBuOE is likely caused by the electrophilic quinone, tBQ (Figure 1A, with the electrophilic moiety denoted in red).
3.5. tBQ Alone Is Less Potent in Cancer Cells than the Combination of tBHQ and MnBuOE
To investigate whether the toxicity of the tBHQ–MnBuOE combination was caused by the electrophilic quinone tBQ, Jurkat and MDA-MB-231 cells were treated with tBQ, or the tBHQ–MnBuOE combination treatment (Figure 5A,B). In both cell lines, tBQ alone was much less toxic than the combination treatment, with an LC50 of 29.1 ± 2 µM for Jurkat cells and >200 µM for MDA-MB-231 cells. The combination of tBHQ and MnBuOE was far more potent, with LC50 values of 4.3 ± 0.2 µM and 23.9 ± 1 µM for Jurkat and MDA-MB-231 cells, respectively. Experiments performed with a different passage number in Jurkat cells showed similar results for tBQ toxicity (Figure S4F). The data suggest that the combination of tBHQ and MnBuOE would likely make a more effective therapeutic than administering tBQ.
3.6. The Selectivity of the Toxicity of the tBHQ–MnBuOE Treatment to Cancer Cell Lines
While the combination of tBHQ and MnBuOE generates levels of oxidative stress that are toxic to the tested cancer lines, primary cells from healthy tissue are typically less sensitive to induced oxidative stress. To test whether primary cells might be less affected by the treatment, tBHQ and MnBuOE were tested in non-cancerous mouse prostate fibroblasts (MPF) and compared to PC3 cells. PC3 cells were sensitive to the tBHQ–MnBuOE treatment as expected, with viability dropping by half at 10 µM tBHQ with MnBuOE present (Figure 6A). MPF cells, on the other hand, were slightly sensitive to 1 μM MnBuOE alone, with 80% viability, but adding tBHQ with MnBuOE did not further decrease viability, up to 20 µM tBHQ (Figure 6B). This finding that cancer cell lines are more sensitive to tBHQ and MnBuOE than MPF supports a potential therapeutic usage for this combination of antioxidants.
Use of dtBHQ to probe the mechanism of cell death suggests that oxidative stress is not due to tBHQ redox cycling. (A) UV–Vis spectra of the tBHQ–MnBuOE reaction. The dashed line is the spectrum of the tBHQ–MnBuOE mixture immediately after mixing, and the solid line is the reaction near completion (22 min). (B) UV–Vis spectra of the tBHQ–MnBuOE reaction. The dashed line is the spectrum of the dtBHQ–MnBuOE mixture immediately after mixing, and the solid line is the reaction near completion (42 min). (C) The viability of PC3 cells after addition of dtBHQ alone or in combination with MnBuOE for 24 h, as measured by CTGlo assay. Viability was normalized to the untreated control. n = 5 (technical replicates). (D,E) Mitochondrial superoxide was measured in PC3 cells by the MitoSOX assay. Cells were treated with dtBHQ and MnBuOE alone or in combination. n = 2 biological replicates with a total of 5–9 technical replicates. (F,G) In the same treatments as for (C,D), apoptosis or necrosis in PC3 cells was measured by the Annexin V/RedDot assay. n = 2 biological replicates with a total of 3–5 technical replicates. “ns” in the figure indicates a p value that was not significant.
tBQ is less potent than the tBHQ–MnBuOE combination in two cancer cell lines. Cell death from treatment with tBQ was measured in (A) Jurkat cells by the CTF assay and (B) MDA-MB-231 cells by the BCA assay (B). For each condition, viability was normalized to the signal for the lowest concentration of treatment. n = 6 (technical replicates).
tBHQ induces toxicity only in MnBuOE-treated PC3 cells and not in MnBuOE-treated primary cells. Viability upon treatment with tBHQ alone or in combination with MnBuOE for (A) PC3 cells or (B) MPF cells, measured by the Trypan blue exclusion test. n = 3 (biological replicates).
4. Discussion
4.1. tBQ as the Causative Agent of Oxidative Stress and Cytotoxicity
The electrophilic nature of tBQ appears to be largely responsible for the toxic levels of hydrogen peroxide by the tBHQ–MnBuOE treatment, given the lack of toxicity when treating with dtBHQ–MnBuOE instead (Figure 4). This is consistent with the general understanding that arylating (electrophilic) quinones like tBQ are much more toxic than non-arylating quinones that are sterically blocked from acting as electrophiles [30].
Although the dtBHQ experiments suggest tBQ is the agent responsible for oxidative stress and toxicity, tBQ alone was far less potent than the tBHQ–MnBuOE combination (Figure 5). Prior to entering the cell, electrophilic tBQ is known to react with nucleophiles in the cell culture media, such as cysteines in bovine serum album, during the treatment incubation [31]. Of particular relevance to in vivo studies, tBQ would likely react with glutathione in the cytoplasm prior to reaching sensitive organelles such as the ER or the mitochondria [32]. The combination of tBHQ with a manganese porphyrin in therapies may allow for more targeted delivery of tBQ both to the tumor environment and specific organelles. Manganese porphyrins tend to accumulate in mitochondria given that they are highly positively charged and have significant lipophilicity from their alkyl side chains [17].
4.2. How Might tBQ Be Generating Hydrogen Peroxide Through Its Electrophilic Functionality?
The two major sources of reactive oxygen species (ROS) in the cell are the mitochondria and the endoplasmic reticulum. While the electron transport chain is a well-known source of superoxide, disulfide bond formation in the ER also uses molecular oxygen as the terminal electron acceptor, leading to superoxide formation [33]. tBQ is an arylating quinone, a soft electrophile that reacts with the soft nucleophilic cysteine residue in proteins and glutathione. A seminal study by Wang et al. in 2006 demonstrated that arylating quinones form Michael adducts with protein thiols in the endoplasmic reticulum, disrupting disulfide bonds essential for proper protein folding, and triggering the unfolded protein response (UPR) through activation of the protein kinase RNA-like ER kinase (PERK) signaling pathway [30]. In contrast, non-arylating quinones, which lack this electrophilic reactivity, induced significantly less ER stress and toxicity. This result is consistent with our observations that tBHQ, but not dtBHQ, is toxic in combination with MnBuOE (Figure 4, Figures S4E and S5). There is a complex interplay between ER stress and oxidative stress, recently reviewed in [34], but it is clear that ER stress alone is sufficient to generate ROS, shown for example in cells treated with tunicamycin, which disrupts protein folding by blocking N-linked glycosylation [35]. Very recently, Tao et al. reported that PERK disrupts ER–mitochondria communication at mitochondria-associated membranes (MAMs), impairing calcium and lipid exchange and destabilizing mitochondrial homeostasis, leading to the accumulation of mitochondrial ROS [36]. Additionally, numerous studies show mitochondrial oxidative stress generation by electrophiles. For example, the electrophile sulforaphane generates mitochondrial oxidative stress that is responsible for cancer cell death [37,38].
The mechanisms of toxicity of tBQ administered to cultured cells in media have been investigated [32] and in this context, ER stress was not observed. Rather, cellular glutathione levels were depleted, presumably by the export of the tBQ–glutathione conjugate from the cell, and glutathione depletion would generate oxidative stress. However, it seems that under these conditions of total glutathione depletion induced by tBQ added to cell culture media, many other cellular thiols would also be modified by tBQ, leading to general toxicity that would be unlikely to be rescued by catalase as effectively as the tBHQ–MnBuOE treatment-induced toxicity in our study. Collectively, these observations suggest that depletion of total glutathione by conjugation with tBQ is not the source of the toxic levels of hydrogen peroxide from the combination treatment. It remains to be tested whether the combination treatment generates tBQ intracellularly, to help explain differences between extracellular tBQ addition and the tBHQ–MnBuOE combination.
Quinones such as tBQ are well-known to generate toxic levels of hydrogen peroxide in the cell by one-electron redox cycling [39,40]. Quinones are readily reduced by one electron by cytochrome P450 enzymes to unstable semiquinone radical anions, with the electron then being readily donated back to molecular oxygen to generate superoxide and, subsequently, hydrogen peroxide. A major role as an antioxidant enzyme has been identified for NAD(P)H:quinone oxidoreductase (NQO1) to prevent one-electron redox cycling by rapidly reducing quinones back to the hydroquinone (Figure 1A) [41]. Aldo-keto reductase family 1 member B1 (AKR1B1) can reduce tBQ to tBHQ [42]. Both enzymes are widely expressed in various tissues and organs, primarily in the cytoplasm. Reduction in tBQ in the cytoplasm by a relevant reductase may be one reason why tBQ is so much less toxic as a cellular treatment than the tBHQ–MnBuOE combination, which may be able to generate tBQ within particularly vulnerable cellular compartments such as the endoplasmic reticulum and mitochondria.
Given that the dtBHQ–MnBuOE combination treatment is non-toxic (e.g., Figure 4), it seems unlikely that toxic levels of hydrogen peroxide from the combination treatment would be generated by either one-electron or two-electron redox cycling of tBHQ, apart from the electrophilic character of tBQ. dtBHQ is easily oxidized to dtBQ when catalyzed by MnBuOE (Figure 4B), and yet the generated dtBQ does not induce detectable mROS or cytotoxicity. For tBQ and dtBQ generated intracellularly to both undergo one-electron redox cycling, they would be reduced by cytochrome P450s, which seems likely given the high structural diversity among quinones able to generate oxidative stress in this manner [40]. Similarly, for tBQ and dtBQ to both undergo two-electron redox cycling, they would be reduced by cellular reductases such as NQO1, which also seems likely given the structural diversity in the many known substrates of the enzyme [43]. The ability of NQO1 and cytochrome P450s to act on tBQ and dtBQ as substrates remains to be determined.
In sum, the literature suggests that the hydrogen peroxide-dependent toxicity of the combination treatment of tBHQ and a manganese porphyrin, with no observed toxicity from a similar combination treatment of dtBHQ and a manganese porphyrin, is due to ER stress caused by intracellularly generated tBQ. Mechanisms involving one-electron or two-electron redox cycling-generated hydrogen peroxide are less likely.
4.3. Role of the Manganese Porphyrin in Toxicity of the Combination Treatment to Cancer Cells
Previous research in the literature points to a mechanism by which the manganese porphyrin would significantly contribute to the toxicity of the combination treatment. First, intravenous administration of decagrams of ascorbate alone has shown substantial promise in clinical trials [44,45], due the ability of ascorbate to redox cycle and generate levels of hydrogen peroxide that are toxic to cancer cells. There are sufficient free transition metal levels in the cell to catalyze oxidation and thus redox cycling of ascorbate. The inclusion of manganese porphyrins can significantly increase the toxicity of hydrogen peroxide generated by ascorbate toward cancer cells ([46,47], reviewed in [19]). Not only does MnBuOE catalyze redox cycling of ascorbate, producing more hydrogen peroxide, but at high enough ascorbate levels, sufficient hydrogen peroxide is generated to switch MnBuOE to an “oxidative” function. As largely discovered by the Tome lab, sufficient concentrations of hydrogen peroxide oxidize manganese porphyrins from the +5 to the +3 state, which then catalyzes the oxidation of protein thiols inducing S-glutathionylation, causing massive changes to signaling and function in the cell, as well as overall oxidation of the glutathione pool. Healthy cells, by comparison, have sufficient antioxidant enzyme capacity, induced largely by the Nrf2 transcription factor in response to oxidative stress, to keep hydrogen peroxide levels below the threshold that can oxidize manganese porphyrins to the +3 state. This capacity of normal cells explains why a combination treatment of a manganese porphyrin with a hydrogen peroxide-generating molecule shows selective toxicity to cancer cells. Importantly, inclusion of catalase in the media indicated that the toxicity of an ascorbate–manganese porphyrin combination was completely dependent on hydrogen peroxide in breast cancer cell lines SUM149 [46] and MDA-MB-231 [47], as was observed in this study for the toxicity of the tBHQ–MnBuOE combination. Thus, our current working model is that the tBHQ–MnBuOE combination generates sufficient hydrogen peroxide to oxidize MnBuOE to the +3 state, followed by an increase in the glutathione disulfide (GSSG)/GSH ratio, S-glutathionylation of a broad swath of cellular proteins, and apoptotic cell death (Figure 7). The concentration of tBHQ needed in combination with a manganese porphyrin to kill cancer cells is much lower than the concentration of ascorbate required for a similar effect. This difference in LC50 may arise because ascorbate generates hydrogen peroxide through redox cycling, whereas we hypothesize that tBHQ produces hydrogen peroxide via a different mechanism—likely ER stress—through its electrophilic quinone form, tBQ.
Supporting the model of the “oxidant” function of manganese porphyrins, oxidation to the +3 state, being enabled by tBQ produced in the tBHQ–MnBuOE combination (Figure 7), we have previously shown that 50 µM dtBHQ in HaCaT cells suppressed global protein synthesis but did not generate enough hydrogen peroxide to activate the “oxidant” function of MnTMPyP [49]. Instead, MnTMPyP rescued cells from protein synthesis inhibition induced by dtBHQ, presumably by acting in its superoxide dismutase capacity. In contrast, adding MnTMPyP to HaCaT cells treated with tBHQ increased toxicity (Figure S1). Because dtBHQ induces oxidative stress sufficient to inhibit protein synthesis, yet MnTMPyP reverses this effect rather than causing toxicity, it suggests that dtBHQ-driven redox cycling produces hydrogen peroxide at levels too low to oxidize manganese porphyrin to the +3 state. Indeed, 50 µM dtBHQ did not alter the [GSH]:[GSSG] ratio in HaCaT cells [23], even though global protein synthesis was suppressed at that concentration [49].
In sum, findings from this work and the literature support a working model (Figure 7) in which the manganese porphyrin plays two critical roles—first, in its +5 state, catalyzing tBHQ to tBQ (which then induces the production of hydrogen peroxide), and second, in its +3 state induced by high concentrations of hydrogen peroxide, catalyzing the oxidation of GSH and S-glutathionylation of cellular thiols.
4.4. Therapeutic Potential of a tBHQ-Manganese Porphyrin Combination
The apparent selectivity of the tBHQ–MnBuOE combination for cancer cells, LC50 values that are likely achievable physiologically, and the use of tBHQ as a common food preservative and MnBuOE in clinical trials as a radioprotectant together make this finding an exciting possible avenue for cancer treatment. An important next step prior to animal studies is to test an array of primary cells from healthy tissue for possible susceptibility. In addition, other manganese porphyrins in clinical development display varying degrees of targeting to the tumor environment and cellular organelles such as the mitochondria [17]. These capabilities may enhance selective production of tBQ from tBHQ in vulnerable cellular compartments in cancer cells, widening the therapeutic window of a combination therapy.
5. Conclusions
The combination of tBHQ and MnBuOE, each primarily known for their antioxidant effects, unexpectedly causes oxidative stress and apoptotic cell death in cancer cells. This oxidative stress is likely a result of the ability of the manganese porphyrin to catalyze the oxidation of tBHQ to tBQ inside the cell. The electrophilic nature of tBQ plays an essential role in generating levels of hydrogen peroxide in the tBHQ–MnBuOE combination treatment that are toxic to cancer cells. According to current knowledge in the field, ER stress is a likely means by which the generated tBQ produces hydrogen peroxide in these cells, potentially both from the ER itself and from mitochondria. The amount of hydrogen peroxide produced is likely sufficient to interact with the manganese porphyrin and cause extensive S-glutathionylation of cellular targets, a well-described mechanism for toxicity caused by manganese porphyrins in combination with hydrogen peroxide.
6. Patents
Eggler, A.L.; Tamarin, S.; Biesterveld, L.; LaMorte, J. Pharmaceutical composition comprising oxidizable biphenol and manganese porphyrin, and method of using the same. U.S. Patent No. 12201649. 2025. U.S. Patent and Trademark Office.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Chen S. Cao Z. Prettner K. Kuhn M. Yang J. Jiao L. Wang Z. Li W. Geldsetzer P. Bärnighausen T. Estimates and Projections of the Global Economic Cost of 29 Cancers in 204 Countries and Territories From 2020 to 2050 JAMA Oncol.2023946547210.1001/jamaoncol.2022.782636821107 PMC 9951101 · doi ↗ · pubmed ↗
- 2Lowenthal R.M. Eaton K. Toxicity of Chemotherapy Hematol. Oncol. Clin. North Am.19961096799010.1016/S 0889-8588(05)70378-68811311 · doi ↗ · pubmed ↗
- 3Zahavi D. Weiner L. Monoclonal Antibodies in Cancer Therapy Antibodies 202093410.3390/antib 903003432698317 PMC 7551545 · doi ↗ · pubmed ↗
- 4Zhao L. Cao Y.J. Engineered T Cell Therapy for Cancer in the Clinic Front. Immunol.201910225010.3389/fimmu.2019.0225031681259 PMC 6798078 · doi ↗ · pubmed ↗
- 5Zugasti I. Espinosa-Aroca L. Fidyt K. Mulens-Arias V. Diaz-Beya M. Juan M. Urbano-IspizuaÁ. Esteve J. Velasco-Hernandez T. Menéndez P. CAR-T Cell Therapy for Cancer: Current Challenges and Future Directions Signal Transduct. Target. Ther.20251021010.1038/s 41392-025-02269-w 40610404 PMC 12229403 · doi ↗ · pubmed ↗
- 6Hernandez I. Samuel W. Bott B.S. Anish S. Patel B.S. Collin G. Wolf B.S. Alexa R. Hospodar B.S. Shivani Sampathkumar B.S. Pricing of Monoclonal Antibody Therapies: Higher If Used for Cancer?Am. J. Manag. Care 20182410911229461857 · pubmed ↗
- 7Di M. Potnis K.C. Long J.B. Isufi I. Foss F. Seropian S. Gross C.P. Huntington S.F. Costs of Care during Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory B-Cell Lymphomas JNCI Cancer Spectr.20248 pkae 05910.1093/jncics/pkae 05939115391 PMC 11340641 · doi ↗ · pubmed ↗
- 8Du J. Cullen J.J. Buettner G.R. Ascorbic Acid: Chemistry, Biology and the Treatment of Cancer Biochim. Biophys. Acta BBA Rev. Cancer 2012182644345710.1016/j.bbcan.2012.06.003PMC 360847422728050 · doi ↗ · pubmed ↗