Mas-Related G-Protein-Coupled Receptors: Emerging Roles in Neuropathic Pain
Mario García-Domínguez

TL;DR
This paper reviews the role of Mas-related G-protein-coupled receptors in neuropathic pain and their potential as drug targets.
Contribution
The paper highlights novel insights into how these receptors modulate pain hypersensitivity and their therapeutic potential.
Findings
Mas-related GPCRs influence neuronal excitability and inflammatory responses in neuropathic pain.
These receptors interact with ion channels and immune mediators to regulate pain signaling.
Targeting specific receptor subtypes may lead to new analgesic therapies.
Abstract
Mas-related G-protein-coupled receptors constitute a distinct family of GPCRs expressed in some subsets of sensory neurons and immune cells. Increasing evidence highlights their contribution to the modulation of nociceptive signaling and neuroimmune interactions. Recent studies demonstrate that Mas-related G-protein-coupled receptors are implicated not only in itch transmission but also in the pathophysiology of neuropathic pain, where aberrant receptor activity influences neuronal excitability, glial activation, and inflammatory responses. This review summarizes current knowledge on the molecular mechanisms by which Mas-related G-protein-coupled receptors regulate pain hypersensitivity, including their interactions with ion channels, neuropeptides, and immune mediators. Moreover, the potential of targeting specific Mas-related G-protein-coupled receptor subtypes for therapeutic…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
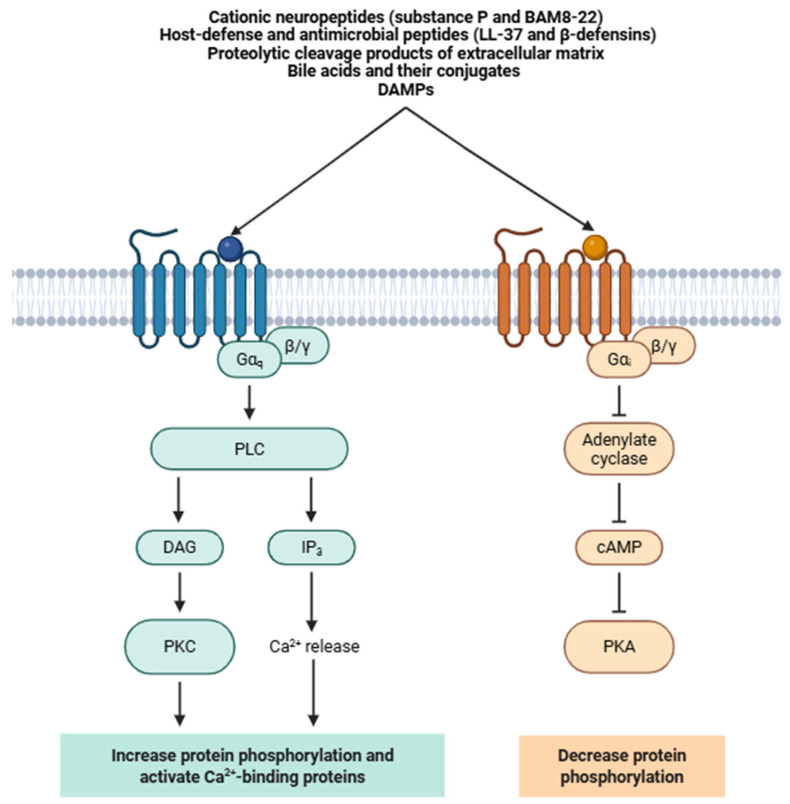 Figure 1
Figure 1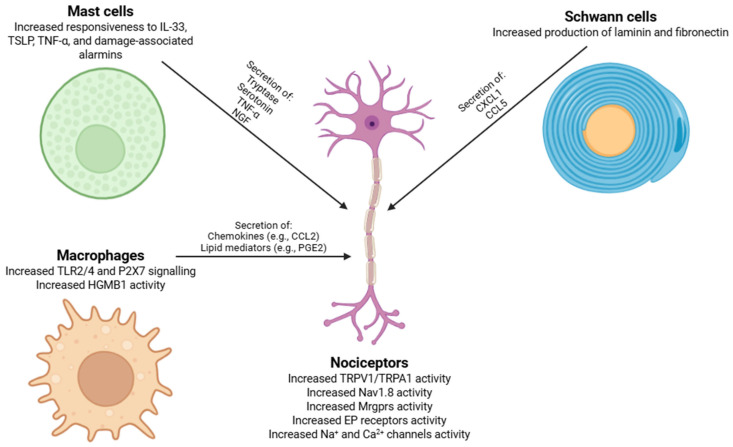 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMast cells and histamine · Nicotinic Acetylcholine Receptors Study · Receptor Mechanisms and Signaling
1. Introduction
According to the International Association for the Study of Pain (IASP), pain is defined as “an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage” [1,2]. Pain is highly characterized as a multifactorial and inherently multidimensional phenomenon, acting as a critical diagnostic marker and encompassing sensory-discriminative, affective-emotional, and cognitive-evaluative dimensions, frequently indicating underlying health conditions that necessitate medical evaluation [3,4]. Pain can be divided into three principal types according to the causative agent. Nociceptive pain arises from real or potential damage to non-neural tissue through nociceptor activation [5], whereas nociplastic pain results from altered nociception without evident tissue injury or detectable somatosensory lesions [6].
Neuropathic pain is a chronic, prevalent, and disabling condition affecting approximately 7–10% of the population [7,8,9]. Its etiology is highly heterogeneous, encompassing traumatic nerve injury, chemotherapy-induced neuropathy (CIPN), numerous metabolic disorders (e.g., diabetes), viral infections (e.g., HIV/AIDS or herpes zoster), autoimmune neuropathies (e.g., Guillain-Barré syndrome -GBS-), spinal cord injury, and neurodegenerative diseases (e.g., multiple sclerosis) [10,11,12,13,14,15,16]. Clinically, neuropathic pain manifests as a constellation of positive (e.g., spontaneous burning pain and electric-shock-like sensations) and negative (e.g., loss of sensory function) symptoms, as well as evoked phenomena such as allodynia (pain in response to non-noxious stimuli) and hyperalgesia (exaggerated pain to noxious stimuli) [17]. Despite this heterogeneity, a defining feature of neuropathic pain is persistent neuroplasticity across numerous levels of the nociceptive pathway, encompassing peripheral sensitization, ectopic activity in injured sensory neurons, maladaptive neuron-immune interactions, synaptic remodeling in the spinal dorsal horn, and dysregulated descending pain modulation [18,19,20]. In terms of molecular pathways, peripheral or central injury triggers the release of numerous pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6), chemokines (e.g., CCL2, CCL3, and CCL5), and damage-associated molecular patterns (DAMPs) [21,22,23]. Moreover, activated glial cells further release pro-inflammatory mediators and neurotrophic factors (e.g., IL-1β, TNF-α, BDNF, and NGF), while chemokine gradients recruit peripheral immune cells to injured nerves and central pain-processing regions [24,25]. These processes induce alterations in several ion channels (e.g., VGSCs, VGCCs, and K^+^ channels) and neurotransmitter systems, thus promoting neuronal hyperexcitability [26,27,28].
Current therapeutic strategies remain limited in both efficacy and tolerability. First-line pharmacotherapies (such as gabapentinoids, tricyclic antidepressants -TCAs-, serotonin-noradrenaline reuptake inhibitors -SNRIs-, and numerous sodium channel blockers) produce clinically relevant pain reduction in approximately 30% of patients [29,30]. Opioids show reduced long-term effectiveness in neuropathic conditions and introduce risks of dependence, tolerance, respiratory depression, and opioid-induced hyperalgesia [31]. Even emerging approaches such as monoclonal antibodies or neurostimulation have not fully addressed the critical need for mechanism-based, peripherally selective analgesics [32,33]. This therapeutic stagnation highlights the need to identify novel molecular targets capable of modulating pathological pain signals without disrupting normal nociceptive function or causing systemic side effects.
One such group of targets is the family of G-protein-coupled receptors (GPCRs), the largest and pharmacologically most exploited receptor class in the human genome [34]. GPCRs act as molecular transducers that convert extracellular chemical signals into intracellular second-messenger cascades, thus regulating neuronal excitability, synaptic plasticity, neurotransmitter release, and neuroimmune interactions [35]. Many of the most effective analgesic drugs currently in clinical use (including μ-opioid receptor agonists, cannabinoid receptor agonists, and α2-adrenergic receptor ligands) achieve their actions via GPCR signaling pathways, demonstrating the druggability of this receptor family [36].
Within this superfamily, the Mas-related G-protein-coupled receptors (Mrgprs) have emerged as a particularly compelling subset due to their restricted expression in small-diameter nociceptors of dorsal root (DRG) and trigeminal ganglia and their unique pharmacological profiles [37,38]. Firstly, classified as orphan receptors, Mrgprs are now known to respond to a diverse spectrum of ligands, like endogenous amino acids and neuropeptides (e.g., BAM8-22, β-alanine, and substance P), inflammatory mediators, and some exogenous xenobiotics [39,40]. Functionally, several Mrgpr subtypes (principally MrgprA3, MrgprC11, and the human ortholog MRGPRX1) have been implicated in both pruriceptive and nociceptive signaling [41,42,43,44,45]. Their activation initiates canonical Gαq/11-phospholipase C (PLC) pathways, leading to IP_3_ formation, intracellular Ca^2+^ mobilization, and downstream protein kinase C (PKC)-mediated sensitization of transient receptor potential (TRP) channels, like TRPA1 and TRPV1 [45,46,47]. Alongside neuronal activation, Mrgprs participate in bidirectional mast cell-neuron signaling, modulate cytokine and neuropeptide release, and influence glial reactivity in the dorsal horn during chronic pain [48,49,50,51].
Current evidence highlights Mrgprs as fundamental molecular nodes connecting peripheral damage, inflammatory signaling, and neuronal hyperexcitability, underlying the chronicity of neuropathic pain [52,53]. Their distribution, ligand diversity, and mechanistic involvement in pathological but not baseline nociception make them a promising, yet still underexploited, class of therapeutic targets [54]. A more comprehensive understanding of Mrgpr signaling might enable the development of next-generation analgesics with improved selectivity, reduced central side effects, and minimal addiction liability [50].
This review synthesizes current knowledge of Mrgprs and their key roles in the pathophysiology of neuropathic pain, a condition whose high prevalence and limited responsiveness to existing therapies make it an ideal context for investigating Mrgprs as potential targets for next-generation, mechanism-based analgesics. The molecular mechanisms by which Mrgprs modulate nociceptor excitability, neuropeptide release, and neuroimmune communication are examined, highlighting their contribution to persistent pain and hypersensitivity. Preclinical and translational evidence support Mrgprs as promising targets for next-generation analgesics.
2. The Mas-Related G-Protein-Coupled Receptor Family
2.1. Mrgpr Classification and Subtype Diversity
Mrgprs represent a discrete subfamily of class A GPCRs distinguished by extensive evolutionary diversification and a predominantly peripheral expression pattern [55]. In contrast to canonical GPCR families that exhibit high cross-species sequence conservation, the Mrgpr repertoire displays substantial lineage-specific expansion, mainly in rodents, suggesting adaptive evolution driven by pressures in sensory and host-defense pathways [56]. Mrgprs were initially identified through transcriptomic analyses of DRG, where they were characterized as orphan GPCRs selectively expressed in subsets of nociceptive and pruriceptive neurons [57,58]. Since then, this family has expanded to include many receptors with divergent ligand selectivity, tissue distribution, and physiological roles [50].
In mice, the Mrgpr gene family is represented by more than 50 genes, many located in two large clusters on chromosome 7 [50]. These receptors are classified into the Mrgpra, Mrgprb, Mrgprc, Mrgprd, Mrgpre, Mrgprf, Mrgprg, and Mrgprx subfamilies (Table 1), although the boundaries between subgroups are based primarily on sequence similarity rather than strict phylogenetic lineage [50], and several murine receptors lack direct human orthologs, complicating translational research by limiting direct cross-species extrapolation of pharmacological findings [39]. The human genome, in contrast, contains a very reduced number of functional Mrgpr genes, such as MRGPRX1-4, MRGPRD, MRGPRE, MRGPRF, and a small number of additional loci (Table 1) [40], and this contraction of the gene family in primates is not indicative of functional loss but rather reflects a shift toward receptor specialization instead of multiplicity [55].
Overall, Mrgpr classification is an evolving framework informed by comparative genomics, structural studies, and functional de-orphanization. Once considered a niche sensory receptor group, Mrgprs are now recognized as key molecular mediators at the neuro-immune interface, with ongoing single-cell transcriptomics, humanized mouse models, and structure-guided ligand discovery poised to further refine their taxonomy and functional roles [46,59,60].
2.2. Molecular Signaling Pathways
A key molecular characteristic of Mrgprs is their divergence from several highly conserved motifs present in canonical class A GPCRs. Unlike prototypical class A receptors, many Mrgpr subtypes, such as MrgprA3, MrgprC11, and MrgprD, do not reserve the canonical DRY motif (Asp-Arg-Tyr) at the cytoplasmic end of TM3 or the NPxxY motif (Asp-Pro-x-x-Tyr) on TM7, motifs that function as microswitches for activation and G protein coupling, rather than directly defining the orthosteric binding site [39,46].
Independent of these microswitch motifs, sequence comparisons across Mrgpr subfamilies also reveal reduced conservation of several residues that conventionally constitute the orthosteric binding pocket [46]. In Mrgprs, these positions frequently exhibit substitutions to residues with divergent side chains (e.g., non-polar or small aliphatic residues in place of conserved polar/aromatic residues), and the overall pattern of side-chain chemical landscape of the pocket is heterogeneous across the MrgprA, MrgprB, and MrgprC clades [46,61,62].
Endogenous agonists of Mrgprs encompass an exceptionally diverse array of molecular species (Figure 1), including cationic neuropeptides (e.g., substance P and BAM8-22) [63], host-defense and antimicrobial peptides (e.g., LL-37 and β-defensins) [64], proteolytic cleavage products of extracellular matrix [65], bile acids and their conjugates [66], and DAMPs [55]. Exogenous ligands include a wide range of structurally unrelated small molecules, including anesthetics [67], neuromodulatory drugs [68], opioids [68], and environmental irritants [69], many of which act as biased agonists or partial agonists depending on the receptor subtype and cellular context. A subset of Mrgpr paralogs remain orphan receptors, whose cognate ligands have yet to be identified [70].
Finally, while this structural divergence in the putative ligand-facing residues is consistent with functional studies showing that different Mrgpr subtypes can be activated by chemically distinct ligands, direct biophysical evidence linking these specific residue differences to broad “promiscuity” in ligand binding is currently limited. Consequently, rather than describing Mrgprs as inherently promiscuous, a more precise interpretation is that divergence from conserved orthosteric pocket residues contributes to the diversification of ligand sensitivity profiles across Mrgpr subfamilies, in a ligand class- and receptor subtype-dependent manner [61].
At the signaling level, most members of the Mrgpr family preferentially couple to the Gαq/11 subfamily of heterotrimeric G proteins (Figure 1) [71]. Upon agonist engagement, conformational rearrangements within the receptor (principally rotational and outward movements of transmembrane helices 5, 6, and 7) stabilize an active-state conformation that evokes GDP-GTP exchange on the Gαq/11 subunit [44]. This nucleotide exchange triggers the dissociation of the Gαq/11-GTP complex from the Gβγ dimer, allowing both signaling entities to interact with distinct downstream effectors. Activated Gαq/11 subsequently engages and activates phospholipase Cβ (PLCβ), which hydrolyzes the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP_2_) into two second messengers: inositol 1,4,5-triphosphate (IP_3_) and diacylglycerol (DAG) [72]. IP_3_ diffuses via the cytosol and binds to many IP_3_ receptors (IP_3_Rs) located on the endoplasmic reticulum membrane, inducing Ca^2+^ efflux from the endoplasmic reticulum into the cytoplasm [73]. The resulting increase in Ca_i_^2+^ triggers various downstream processes, including activation of Ca^2+^-dependent cation channels (TRPA1 and TRPV1), membrane depolarization, and neurotransmitter or neuropeptide release (e.g., substance P and calcitonin gene-related peptide -CGRP-) [48].
Beyond canonical Gαq/11 coupling, several Mrgpr subtypes exhibit coupling promiscuity or context-dependent signaling plasticity. Depending on receptor subtype, ligand bias, and cellular milieu, some Mrgprs can alternatively engage Gαi/o proteins or β-arrestin-dependent scaffolding pathways [55]. Coupling to Gαi/o can lead to inhibition of adenylyl cyclase, resulting in reduced cyclic AMP (cAMP) levels and modulation of protein kinase A (PKA)-dependent signaling [72]. In contrast, β-arrestin recruitment not only contributes to receptor internalization and desensitization but can also scaffold mitogen-activated protein kinase (MAPK) cascades, including ERK1/2 and p38 MAPK pathways [74]. β-Arrestin-mediated signaling, as observed for MRGPRX2 and MRGPRX4 in mast cells and dependent on β-arrestins 1 and 2, is usually spatially and temporally distinct from G protein-dependent pathways, potentially giving rise to compartmentalized downstream effects [75,76,77].
2.3. Characterized Physiological and Pathophysiological Actions of Mrgprs
Rapid advances in single-cell transcriptomics, in vivo Ca^2+^ imaging, ligand screening technologies, and structural biology have expanded the conceptual framework of Mrgpr biology [78,79]. It is now evident that Mrgprs are not only determinants of sensory neuron identity but also central nodes in intercellular communication between neurons, immune and epithelial cells, and microbiota-derived environmental signals [80,81,82]. Their biological diversity reflects both evolutionary pressures and molecular specialization, resulting in unique patterns of expression, ligand selectivity, and intracellular signaling architecture unparalleled among GPCR families [37,38].
In sensory neurons, Mrgprs (e.g., MrgprA3, MrgprC11, and MrgprD) define distinct pruriceptive and polymodal sensory populations with unique molecular signatures and central projection patterns [47,72,83,84]. MrgprA3 marks a genetically defined lineage that constitutes one of the principal pathways for non-histaminergic itch, responding robustly to chloroquine, SLIGRL peptides, and potentially endogenous pruritogenic metabolites generated under pro-inflammatory conditions [83,84,85]. MrgprC11 shows a broader ligand range, responding to proenkephalin-derived peptides (e.g., BAM8-22), as well as neuropeptides and cytokine-associated factors released during tissue stress [86,87]. MrgprD labels a separate mechanosensitive population implicated in punctate mechanical nociception, environmental toxin detection, and metabolic chemosensation [88,89].
On the other hand, pathological dysregulation of Mrgprs contributes to chronic pruritic, inflammatory, and hypersensitivity conditions [47]. Overactivity of MrgprA3- and MrgprC11-positive neurons is a defining feature of atopic dermatitis, prurigo nodularis, and systemic pruritus [90,91]. These neurons experience transcriptional reprogramming during chronic inflammation, leading to increased receptor expression, heightened excitability, and enhanced responsiveness to pruritogenic mediators [47]. The resulting feed-forward neuroimmune loops perpetuate persistent itch, scratching-induced barrier disruption, and secondary infections that exacerbate disease severity [92].
In non-neuronal tissues, Mrgprs serve roles of comparable significance. MRGPRX2, prominently expressed on human skin- and tissue-resident mast cells, operates as a crucial hub for IgE-independent mast-cell activation [93,94,95]. Its ability to detect several cationic amphipathic peptides (e.g., LL-37, β-defensins, substance P, neurotensin, and vasoactive intestinal peptide) allows it to integrate neural, epithelial, and microbial signals into coordinated inflammatory responses [96,97]. Some small-molecule drugs such as fluoroquinolones, vancomycin, icatibant, neuromuscular-blocking agents, and certain opioids activate MRGPRX2, producing degranulation and mediator release without requirement for prior sensitization [98]. This mechanism underlies a substantial proportion of pseudo-allergic drug reactions characterized by flushing, urticaria, angioedema, bronchospasm, and anaphylactoid events [99]. MRGPRX2-mediated mast cell degranulation releases numerous mediators, including histamine, tryptase, chymase, serotonin, leukotrienes, prostaglandins, and cytokines such as IL-4, IL-6, and TNF-α, thereby generating a potent inflammatory microenvironment capable of modulating neuronal excitability, vascular permeability, and leukocyte recruitment [96,100,101].
In epithelial tissues (e.g., skin, airway mucosa, and gastrointestinal tract), Mrgprs are expressed in keratinocytes, epithelial progenitors, secretory cells, and some subsets of tissue-resident immune cells [101,102]. Their activation controls epithelial barrier integrity, antimicrobial peptide and cytokine secretion, and wound healing responses [102,103,104,105]. In the gastrointestinal system, expression of Mrgpr family members in enteric neurons, enterochromaffin cells, innate lymphoid cells, and smooth muscle fibers contributes to the regulation of motility, visceral sensitivity, mucosal immune responses, and secretomotor function [89,106,107,108]. Mrgpr-mediated detection of some microbial metabolites and dietary compounds may constitute part of a broader neuroimmune surveillance network that orchestrates host-microbe interactions and drives mucosal homeostasis [109,110]. Dysregulation of these pathways has been implicated in conditions such as irritable bowel syndrome, inflammatory bowel disease, and functional dyspepsia, although mechanistic delineation remains an active area of investigation [82,107,111].
The Mrgpr family is definitively a specialized system integrating neuronal, immune, and epithelial signals to regulate sensory processing and neuroimmune communication. The following section summarizes evidence positioning Mrgprs as key regulators in controlling transcriptional reprogramming, ion-channel modulation, and neuroimmune interactions after nerve injury, driving persistent nociceptor hyperexcitability and maladaptive plasticity.
3. Role of Mrgprs in the Pathophysiology of Neuropathic Pain
Members of the Mrgpr family have emerged as important molecular nodes that modulate nociceptor excitability, neuro-immune communication, and maladaptive plasticity following nerve injury. Although originally classified as itch-associated GPCRs, accumulating evidence indicates that numerous Mrgprs participate in pain amplification through finely tuned signaling interactions with several ion channels, neuropeptides, and inflammatory mediators. Neuropathic pain-related dysregulation of Mrgprs reshapes the functional properties of primary sensory neurons and adjacent immune cells, ultimately promoting persistent hyperexcitability and aberrant intercellular communication.
3.1. Transcriptional and Epigenetic Remodeling of Mrgprs After Nerve Injury
Peripheral nerve injury initiates a multilayered molecular cascade that produces a deeply remodeled and pathologically permissive Mrgpr signaling landscape across DRG neurons [37,38] and associated immune populations [80,81,82], ultimately driving persistent nociceptor hyperexcitability. Following axotomy, injury-responsive transcription factors are activated in a temporally structured and mechanistically cooperative sequence. ATF3, induced within hours, binds high-affinity stress-responsive elements embedded in Mrgpr promoters and recruits the histone acetyltransferases CBP and p300, facilitating the deposition of activating chromatin marks such as H3K27ac and H3K9ac and thereby enhancing local chromatin accessibility [112,113,114]. Simultaneously, NF-κB is engaged by TNF-α, IL-1β, and related cytokine signals and, after translocating to the nucleus, removes repressive HDAC complexes from Mrgpr regulatory elements while reinforcing promoter-enhancer looping via interactions with architectural proteins, thereby generating a more permissive and functionally arranged transcriptional microenvironment [115]. On the other hand, STAT3, phosphorylated at Tyr705 in response to IL-6 family cytokines, dimerizes and binds canonical STAT response elements within Mrgpr promoters, recruiting several elongation factors that facilitate productive RNA polymerase II progression and sustain high-amplitude transcription [116,117,118,119]. These transcriptional mechanisms are reinforced by ERK1/2- and JNK-dependent activation of c-JUN, EGR1, and other immediate-early genes, which expand the accessible enhancer landscape in both IB4^+^ non-peptidergic nociceptors and TRPV1^+^/CGRP^+^ peptidergic neurons, promoting the ectopic expression of MrgprA3, MrgprC11, MrgprD, MRGPRX1, and MRGPRX3 in some neuronal subsets that normally lack these receptors [44,84,120,121].
3.2. Mrgpr-Mediated G Protein Signaling and Ion Channel Modulation
Upon ligand engagement, Mrgprs couple to multiple G protein subtypes, integrating Gαq/11-, Gαi/o-, Gαs-, and Gβγ-dependent pathways into a highly convergent modulatory network that alters nociceptor excitability [46,72,122]. Gαq/11 signaling orchestrates PKC-dependent phosphorylation of some channels (Nav1.7, Nav1.8, Nav1.9, TRPV1, and TRPA1), which shifts activation curves leftward, enhances persistent Na^+^ currents, and reduces the threshold for action-potential initiation [123,124,125,126]. Simultaneously, PIP_2_ depletion destabilizes Nav channel gating and blocks KCNQ2/3-mediated M-currents, removing a major constraint on membrane excitability and promoting spontaneous or high-frequency firing [127,128,129].
Mrgpr signaling further releases Gβγ subunits, which strengthen TRPA1 activation kinetics and cooperate with Gαq pathways to streamline microdomain Ca^2+^ signals near active zones, enabling the spatial control of vesicular release and ion channel phosphorylation [130,131]. In neuropathic conditions, Gαi/o pathways that typically inhibit adenylate cyclase can, through injury-induced feedback, engage AC1 to sustain cAMP production and PKA-dependent channel phosphorylation, exacerbating hyperexcitability [132]. β-arrestin scaffolding of ERK1/2 and p38 MAPK links these acute signaling events to longer-term transcriptional reprogramming, including upregulation of ion channel and neuropeptide genes [133]. In addition, specific receptors (e.g., MrgprD) form direct protein-protein complexes with mechanosensitive Piezo2 channels, amplifying the mechanotransduction process and contributing to mechanical allodynia [134].
3.3. Emerging Roles of Mrgprs in Neuroimmune Communication
Peripheral nerve injury engages Mrgprs as crucial organizers of a multilevel neuroimmune circuit that sustains chronic neuropathic hypersensitivity. Peripheral axonal injury induces the recruitment and activation of numerous cell types, including mast cells, macrophages, and Schwann cells, which engage in interconnected reciprocal signaling loops with Mrgpr-expressing nociceptors rather than functioning as isolated effector cells [108].
Some immune cell types undergo coordinated phenotypic remodeling that strengthens neuro-immune communication, and mast cells (Figure 2) in inflamed tissues show elevated MRGPRX2 expression together with an enhanced responsiveness to diverse peptide mediators (IL-33, TSLP, TNF-α, and damage-associated alarmins), leading via Gαq-dependent Ca^2+^ mobilization to exocytosis of histamine-independent mediators such as tryptase, serotonin, TNF-α, and NGF through Rab27b-, Munc13-4-, and SNARE-dependent fusion machinery [135,136,137]. Tryptase induces ERK and PKCε through PAR2, whereas NGF engages TrkA-PI3K/AKT and ERK signaling pathways [138,139]. Collectively, these events enhance TRPV1/TRPA1 activity, increase Nav1.8 function, and upregulate Mrgpr expression on nociceptors, creating a self-reinforcing excitatory state. Similarly, infiltrating macrophages undergo dynamic reprogramming of receptor and cytokine expression in response to interferon signaling, TLR2/4 activation, P2X7 stimulation, and HMGB1, creating an inflammatory milieu that allows engagement of Mrgpr-dependent pathways [48,89]. Macrophage-derived chemokines (e.g., CCL2) and lipid mediators (e.g., PGE_2_) activate EP receptors on Mrgpr^+^ neurons, eliciting PKC- and PKA-dependent phosphorylation of Na^+^ and Ca^2+^ channels, thereby further enhancing neuronal excitability [140,141].
Finally, following peripheral nerve injury, Schwann cells (Figure 2) undergo a phenotype shift from a myelinating to a repair-promoting state. In this context, Schwann cells upregulate the synthesis and secretion of key extracellular matrix proteins, like laminin and fibronectin [142]. Laminin interacts with integrin receptors on the growth cones of regenerating axons, activating intracellular signaling cascades such as the focal adhesion kinase (FAK) and phosphoinositide 3-kinase (PI3K)/Akt pathways, which allow cytoskeletal reorganization and directed axonal elongation [143]. Fibronectin, through binding to α5β1 integrins, further allows adhesion, migration, and branching of regenerating fibers [144]. Moreover, Schwann cells secrete chemokines, such as CXCL1 and CCL5, which act in both autocrine and paracrine manners to modulate axonal growth and attract macrophages that contribute to debris clearance and growth-promoting cytokine release [145].
These injury-induced signals contribute to the upregulation of several Mrgpr family members on regenerating sensory fibers. This upregulation may involve the activation of transcription factors, including c-Jun and STAT3, which bind to the promoter regions of Mrgpr genes, thereby sensitizing regenerating nociceptive neurons and modulating their responsiveness to inflammatory and pruritogenic stimuli during tissue repair [146,147].
3.4. Neuropeptide Release and Peripheral Hyperexcitability
Activation of members of the Mrgpr family in primary afferent nociceptors drives a pronounced Ca^2+^-dependent dysregulation of neurotransmitter and neuropeptide release at peripheral terminals, thereby remodeling the molecular framework of neurogenic inflammation and modulating the synaptic transmission enhancements that underlie neuropathic pain [88,148]. Within this framework, Mrgpr-mediated modulation of presynaptic ion channels and the release machinery synergizes with Nav channel-induced membrane depolarization to augment Ca^2+^ entry, elevating intraterminal Ca^2+^ to supraphysiological concentrations [88,149]. The emergent Ca^2+^ microdomains favor high-affinity synaptotagmin binding, expedite SNARE-complex assembly, and transition synaptic vesicles into a release-ready state, ultimately driving increased exocytosis of substance P, CGRP, galanin, B-type natriuretic peptide (BNP), and other dense-core vesicle cargos far surpassing their typical stimulus-evoked outputs [150,151,152,153]. These neuropeptides, beyond their effects in the dorsal horn, act through their cognate receptors (e.g., NK_1_R, CRLR/RAMP1, and natriuretic peptide receptors) expressed on vascular endothelial cells, perivascular immune cells, and stromal elements of the peripheral inflammatory milieu [154]. Their coordinated activity mediates arteriolar vasodilation, plasma extravasation, endothelial activation, and the directed recruitment of neutrophils, macrophages, and mast cells [155].
Following peripheral nerve injury, Mrgpr^+^ primary afferent nociceptors in laminae I-II of the dorsal horn undergo profound presynaptic and postsynaptic remodeling that potentiates central sensitization [156]. At presynaptic terminals, Cav2.2 (N-type) and Cav3.x (T-type) VGCCs exhibit increased open probability and current density, generating localized Ca^2+^ microdomains that orchestrate synaptotagmin-1/2-mediated vesicle fusion and accelerate SNARE complex assembly (including syntaxin-1, SNAP-25, and VAMP2) at the active zone, with RIM, Munc13, and Bassoon orchestrating vesicle docking and priming to shift the readily releasable pool toward a high-probability release state [157,158,159].
The above-mentioned neurotransmitters couple with postsynaptic NMDA receptors and mGluR5, where activity-dependent phosphorylation by CaMKII, PKC, and Src-family kinases enhances channel open probability, prolongs mean open time, reduces Mg^2+^ block, and increases Ca^2+^ influx, thus driving several downstream signaling cascades [160,161]. Ca^2+^-dependent activation of CaMKII and PKC enables GluA1 Ser831 phosphorylation, promoting AMPA receptor insertion into the postsynaptic density and stabilizing synaptic potentiation [162,163]. Concurrently, ERK1/2 activation and nuclear translocation of phosphorylated CREB enhanced transcription of AMPA (GluA1/2), NMDA (GluN2A/2B) receptor subunits, postsynaptic scaffolds (PSD-95, Homer1b/c, and Shank), and cytoskeletal regulators (cofilin, Rac1, and Arp2/3), resulting in dendritic spine enlargement, increased spine head volume, and stabilization of excitatory synapses [164,165,166].
Finally, excessive glutamatergic and peptidergic input concomitantly diminishes inhibitory synaptic control through retrograde endocannabinoid-mediated suppression of presynaptic release, modulation of GABA/glycine transporter function, and BDNF-driven downregulation of KCC2, thereby generating a state of disinhibition that permits low-threshold mechanoreceptor signals to engage aberrant nociceptive circuits [167,168,169,170,171].
Collectively, Mrgpr activation establishes a feed-forward loop of peripheral and central hyperexcitability that amplifies neurotransmitter and neuropeptide release, remodels synaptic architecture, and promotes maladaptive pain signaling. These insights highlight the therapeutic potential of targeting Mrgprs to disrupt pathological neuropeptide release and hyperexcitability in neuropathic pain.
4. Therapeutic Potential of Targeting Mrgprs
Pharmacological strategies to modulate Mrgprs include the use of agonists, antagonists, and biased ligands. While agonists may engage antinociceptive pathways in a subtype-dependent manner, antagonists are currently the most promising approach to inhibit pain-promoting Mrgpr signaling. Table 2 summarizes those pharmacological agents that are currently under investigation in preclinical models and human studies. This table provides an overview of targeted Mrgpr subtypes and their relevance to pain modulation.
Although the contribution of Mrgpr inhibition has not yet been fully characterized in models of neuropathic pain, accumulating evidence indicates that blockade of multiple Mrgpr subtypes produces robust analgesic effects across a variety of inflammatory, visceral, and pruritic pain models. These findings suggest that Mrgprs play a broader role in nociceptive sensitization and peripheral sensory neuron excitability than previously appreciated. As a result, Mrgpr subtypes represent promising molecular targets for the development of novel analgesic therapies, predominantly in conditions where conventional pain treatments are insufficient or associated with significant adverse effects.
Among the pharmacological agents previously cited, aprepitant merits particular attention, given its molecular mechanism, its relevance to neuropeptide-mediated signaling pathways, and its distinctive translational and clinical significance. Aprepitant blocks substance P interaction with neurokinin-1 receptor (NK_1_R), thereby reducing neuronal excitability, mitigating neurogenic inflammation, and suppressing activation of central emetic pathways in the area postrema and nucleus tractus solitarius [177]. Pharmacokinetically, aprepitant is lipophilic, highly protein-bound (>95%), crosses the blood–brain barrier efficiently, and is mainly metabolized by CYP3A4, with additional minor contributions from CYP1A2 and CYP2C19, conferring clinically relevant pharmacokinetic interaction potential [178].
From a receptor pharmacology perspective, aprepitant has been shown in murine systems to inhibit Mrgprb2 [172]; however, its effects on the human ortholog, MRGPRX2, remain undetermined. Clinically, aprepitant has been established as a selective NK_1_R antagonist via phase II-III randomized controlled trials in chemotherapy-induced nausea and vomiting (CINV), where its central NK_1_R blockade synergizes with 5-HT_3_ receptor antagonists and glucocorticoids to increase response rates in both acute and delayed emetic phases [179]. Beyond its antiemetic indication, several exploratory trials have repurposed aprepitant as an antipruritic agent based on the contribution of substance P-NK_1_R signaling to chronic, largely histamine-independent itch. Some studies, like NCT01683552 [180], NCT02646020 [181], NCT03808805 [182], NCT01963793 [183], and NCT01625455 [184], demonstrated efficacy across non-histaminergic pruritic phenotypes.
Conversely, targeting certain Mrgprs (e.g., MRGPRX2) represents a promising strategy for the prevention of pseudo-allergic reactions and mast cell-driven inflammatory pathologies. However, Mrgprs are involved not only in pathological mast cell degranulation but also in protective immunosurveillance, antimicrobial defense, and the maintenance of tissue homeostasis at barrier interfaces [135,185,186]. As a result, pharmacological antagonism of these receptors may be associated with unintended risks, including impairment of immune responses, disruption of host-microbe interactions, and impaired wound-healing processes [135,185,186].
Collectively, these results highlight the therapeutic potential of simultaneously targeting multiple Mrgpr subtypes to elicit potent analgesic and antipruritic effects, particularly in contexts where conventional therapies are limited or associated with side effects.
5. Conclusions
Mrgprs have shifted from being a poorly characterized subset of sensory receptors to emerging as crucial regulators of nociceptive pathways and fundamental mediators in the development of neuropathic pain. Increasing evidence supports the notion that specific Mrgpr subtypes play a crucial role in both the onset of neuronal sensitization following nerve injury and the persistence of chronic pain. They exert these effects through diverse mechanisms, including modulation of ion channel function, activation of neuroimmune signaling, and facilitation of mast-cell-neuronal communication that sustains persistent hypersensitivity.
Despite considerable progress, our understanding of Mrgpr biology remains limited. Critical knowledge gaps remain concerning the identity and physiological significance of endogenous and exogenous ligands, the subtype-specific signaling pathways activated in distinct neuronal and non-neuronal cell populations, and the degree to which Mrgpr-mediated signaling intersects with canonical nociceptive mechanisms. Resolving these questions is essential for developing a more comprehensive framework of how Mrgprs modulate sensory processing under both physiological and pathological conditions.
Importantly, the restricted and highly patterned expression of Mrgprs within defined subsets of peripheral sensory neurons positions them as compelling targets for next-generation analgesics. By facilitating therapeutic approaches that target discrete molecular nodes specifically linked to pathological pain, rather than inducing widespread neuronal inhibition, Mrgpr-based interventions have the potential to achieve greater efficacy while reducing systemic side effects. In addition, the predominantly peripheral localization of Mrgprs suggests a therapeutic window for modulating nociceptive signaling without substantial CNS engagement, thereby potentially reducing sedation, cognitive impairment, and abuse liability that characterize centrally acting analgesics, particularly opioids. This peripheral selectivity enables the modulation of pain-associated neuroimmune and sensitization pathways that are not effectively targeted by existing therapies, thereby offering a mechanistically distinct and potentially complementary analgesic strategy. On the other hand, this therapeutic potential is underscored by emerging evidence that pharmacological or genetic modulation of Mrgpr activity can attenuate pain behaviors in several preclinical models of neuropathy. Pharmacological and genetic modulation of Mrgpr activity largely converge in showing decreased nociceptive hypersensitivity, supporting a shared causal role in neuropathic pain. However, partial divergence between these approaches has also been observed, likely due to off-target pharmacological effects, developmental compensatory mechanisms in genetic models, cell-type-specific receptor functions, interspecies differences, and ligand-biased signaling mechanisms.
While Mrgprs represent a promising therapeutic target, it should be acknowledged that the translational development of Mrgpr-targeted interventions remains at an early-stage relative to more established analgesic strategies, such as opioids, CGRP modulators, or anti-NGF monoclonal antibodies. Continued preclinical validation, optimization of ligand specificity, and demonstration of efficacy and safety in clinically relevant models will be essential before Mrgpr-based therapies can reach comparable developmental maturity.
Taken together, current evidence underscores the potential of Mrgprs as both mechanistic biomarkers and therapeutic targets in the context of neuropathic pain. Continued investigation into Mrgpr ligand specificity, intracellular signaling mechanisms, and interactions with the neuroimmune environment will not only enhance our understanding of pain pathophysiology but may also drive the development of more selective and effective therapeutic strategies for a condition that remains exceptionally difficult to manage.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Raja S.N. Carr D.B. Cohen M. Finnerup N.B. Flor H. Gibson S. Keefe F.J. Mogil J.S. Ringkamp M. Sluka K.A. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises Pain 20201611976198210.1097/j.pain.000000000000193932694387 PMC 7680716 · doi ↗ · pubmed ↗
- 2Vader K. Bostick G.P. Carlesso L.C. Hunter J. Mesaroli G. Perreault K. Tousignant-Laflamme Y. Tupper S. Walton D.M. Wideman T.H. The Revised IASP Definition of Pain and Accompanying Notes: Considerations for the Physiotherapy Profession Physiother. Can.20217310310610.3138/ptc-2020-0124-gee 34456418 PMC 8370731 · doi ↗ · pubmed ↗
- 3Fashler S.R. Cooper L.K. Oosenbrug E.D. Burns L.C. Razavi S. Goldberg L. Katz J. Systematic Review of Multidisciplinary Chronic Pain Treatment Facilities Pain Res. Manag.20162016596098710.1155/2016/596098727445618 PMC 4904600 · doi ↗ · pubmed ↗
- 4Forte G. Favieri F. De Pascalis V. Casagrande M. To Be in Pain: Pain Multidimensional Questionnaire as Reliable Tool to Evaluate Multifaceted Aspects of Pain J. Clin. Med.202413588610.3390/jcm 1319588639407946 PMC 11477689 · doi ↗ · pubmed ↗
- 5Sima S. Lapkin S. Gan Z. Diwan A.D. Nociceptive pain assessed by the Pain DETECT questionnaire may predict response to opioid treatment for chronic low back pain Heliyon 202410 e 2583410.1016/j.heliyon.2024.e 2583438356562 PMC 10865323 · doi ↗ · pubmed ↗
- 6Kaplan C.M. Kelleher E. Irani A. Schrepf A. Clauw D.J. Harte S.E. Deciphering nociplastic pain: Clinical features, risk factors and potential mechanisms Nat. Rev. Neurol.20242034736310.1038/s 41582-024-00966-838755449 · doi ↗ · pubmed ↗
- 7van Hecke O. Austin S.K. Khan R.A. Smith B.H. Torrance N. Neuropathic pain in the general population: A systematic review of epidemiological studies Pain 201415565466210.1016/j.pain.2013.11.01324291734 · doi ↗ · pubmed ↗
- 8Martins J.P. Marson F.A.L. A narrative review of the complex panorama regarding chronic neuropathic pain mainly for the psychological issues Heliyon 202410 e 3828210.1016/j.heliyon.2024.e 3828239403499 PMC 11472089 · doi ↗ · pubmed ↗