New mitochondrial genomes of Saccostrea (Mollusca, Ostreidae) specimens from Hainan Island
Dansheng Xie, Fengping Li, Mingjie Liu, Likai Fan, Xingchen Guo, Aimin Wang, Chunsheng Liu, Zhifeng Gu, Yi Yang

TL;DR
This study sequenced mitochondrial genomes of oysters from Hainan Island to improve their taxonomic classification.
Contribution
The study provides new mitochondrial genome data and identifies two novel OTUs in the genus Saccostrea.
Findings
The four Saccostrea specimens had conserved mitochondrial gene arrangements with a fragmented 16S rRNA gene.
Two novel OTUs were identified, with one showing high intraspecific divergence.
Publicly available Saccostrea genomes were reclassified into nine OTUs, highlighting mislabeling in GenBank.
Abstract
The taxonomic classification of oysters, particularly within the genus Saccostrea, remains poorly resolved due to limitations in traditional morphological and single-gene approaches. To address this, we sequenced the mitochondrial genomes of four Saccostrea specimens (Saccostrea MBSR 211-214) from Hainan Island. The complete mitochondrial genomes (16,280–16,289 bp) revealed a conserved gene arrangement, including 12 protein-coding genes (PCGs), 23 transfer RNA (tRNA), and two ribosomal RNA (rRNA) genes on a single strand, with a fragmented 16S rRNA gene as a distinctive feature. Different OTU delimitation methods consistently identified these four lineages as two novel operational taxonomic units (OTUs). Within one of the OTUs, there are high levels of intraspecific divergence, exhibiting the potential for independent evolution. Our analysis reclassified the publicly available…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
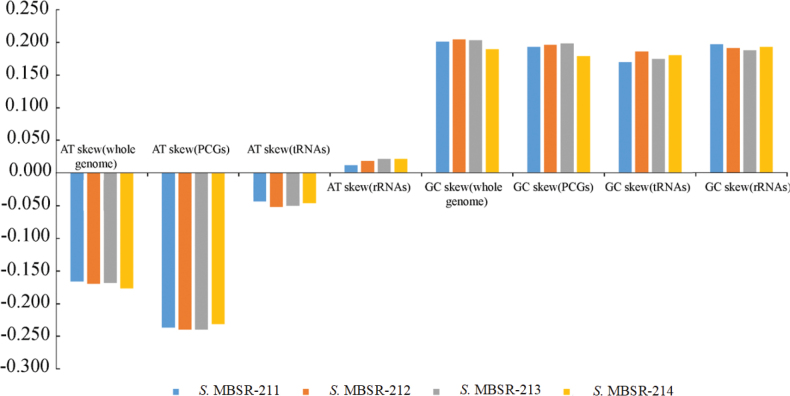 Figure 1
Figure 1 Figure 2
Figure 2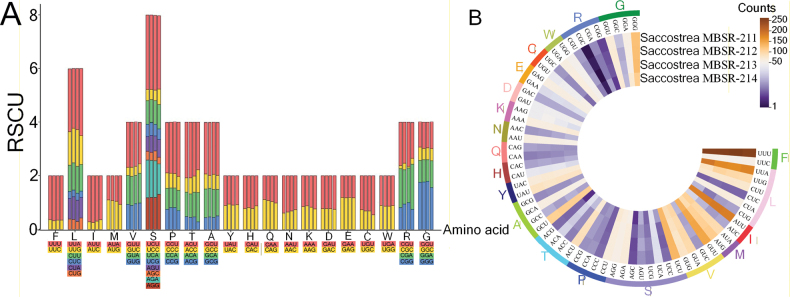 Figure 3
Figure 3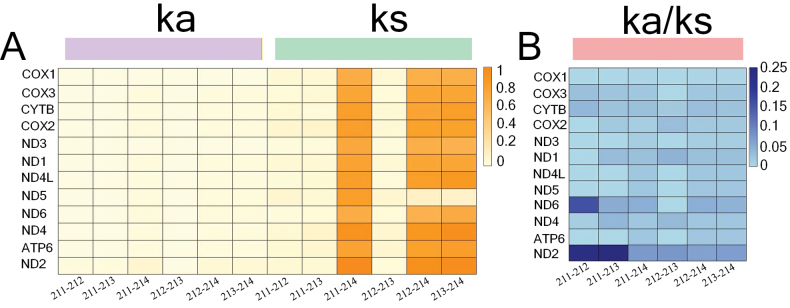 Figure 4
Figure 4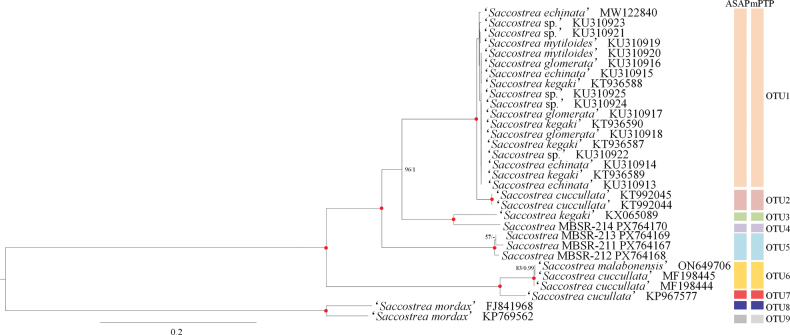 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMarine Bivalve and Aquaculture Studies · Marine Biology and Ecology Research · Genomics and Phylogenetic Studies
Introduction
The taxonomic classification and phylogenetic relationships of oysters remain unresolved, particularly within the genus Saccostrea (Lam and Morton 2006; Sekino and Yamashita 2016; Cui et al. 2021). Traditional morphological approaches have proven problematic, with Stenzel (1971) and Harry (1985) recognizing only a single valid species (S. cucullata) within Saccostrea for the Indo-Pacific region. However, the phenotypic plasticity of Saccostrea varies significantly across different life stages and habitat conditions (Lam and Morton 2006; Reece et al. 2008; McDougall et al. 2024). This plasticity not only leads to substantial intraspecific diversity but also results in extensive interspecific morphological overlap, rendering shell-based taxonomy unreliable (Lam and Morton 2006; McDougall et al. 2024). This complexity is further compounded by frequent interspecific/intraspecific hybridization, introgression events, and the presence of cryptic species complexes, all of which pose additional challenges to conventional morphological identification approaches (Sekino and Yamashita 2013; Hamaguchi et al. 2014; Li et al. 2023). DNA sequencing offers a solution to these limitations, providing more robust evidence for oyster classification (Liu et al. 2011).
The taxonomic complexity of Saccostrea has been investigated through molecular approaches. Lam and Morton (2006) established a reliable and reproducible framework using 16S rRNA data, identifying two major complexes in the Indo-Pacific: the S. cucullata complex (lineages A–G plus S. kegaki and S. glomerata) and the S. mordax complex (lineages A and B). Subsequent studies by Sekino and Yamashita (2013) recognized S. palmula and three S. mordax lineages (A–C) from Okinawa Island. Hamaguchi et al. (2014) revised the taxonomic assignment of S. glomerata to Japanese specimens, asserting that most populations should be classified as S. cucullata, except for S. kegaki, S. mordax, and S. mytiloides and two lineages tentatively assigned to S. malabonensis. The Caribbean populations reveal further complexity (Lohan et al. 2015), while Sekino and Yamashita (2016) identified seven mitochondrial lineages (C, F, G, H, I, J, plus S. kegaki), although only three showed clear reproductive isolation. Li et al. (2017) resolved taxonomic ambiguities by demonstrating conspecificity between Myanmar populations and distinct reference species, advocating for the recognition of S. malabonensis as a valid species. Cui et al. (2021) contributed to this framework by formally describing a new species (S. mordoides) from the S. mordax complex in southern China. Collectively, these studies highlight the limitations of single-gene approaches, with Guo et al. (2018) suggesting climatic oscillations drove recent speciation events in these oyster complexes.
The tropical western Pacific Ocean represents a global biodiversity hotspot for oysters, distinguished by both the youngest fossil records and the highest species richness among extant oyster populations, especially for Saccostrea (Li et al. 2021). Within this biogeographically significant region, Hainan Island’s diverse coastal environments, exceptional water quality, and abundant oyster resources (Lu et al. 2024) provide an ideal natural laboratory for evolutionary studies. A prior DNA barcoding investigation employing COI sequences by Heng et al. (2025) revealed multiple phylogenetically distinct lineages within Saccostrea. These lineages exhibited consistent genetic divergence (typically > 2%) when compared to existing Saccostrea COI sequences in their investigation or deposited in GenBank, suggesting substantial underestimation of the Saccostrea diversity. However, the definitive taxonomic classification of these lineages remained unresolved due to the analytical constraints inherent to single-gene barcoding approaches. Given the inherent limitations of single-gene markers in resolving these issues, advanced molecular genetic tools have become essential for precise species delineation. Oyster mitochondrial genomes, characterized by their rapid evolutionary rates, conserved structural architecture, and strict maternal inheritance pattern, have emerged as particularly valuable molecular markers for systematic studies (Ren et al. 2010; Wu et al. 2010; Yu and Li 2012). The ongoing decline in high-throughput sequencing costs has facilitated a marked increase in the availability of molluscan mitochondrial genome data in recent years. To address this knowledge gap, our study sequenced complete mitochondrial genomes from four Saccostrea specimens collected from Hainan Island. Through integrated analyses of mitochondrial genomic architecture and genus-level phylogenetic reconstruction, we re-evaluate the diversity of Saccostrea and establish the taxonomic validity of these evolutionarily significant specimens.
Materials and methods
Sample collection and DNA extraction
Four specimens representing four Saccostrea individuals (designated as Saccostrea MBSR 211–214) were selected based on the DNA barcoding study by Heng et al. (2025). The specimens were collected from different locations in Hainan Province, China: Saccostrea MBSR-211 from Chengmai (19°59'15"N, 109°53'3"E), Saccostrea MBSR-212 from Danzhou (19°31'15"N, 108°56'58"E), Saccostrea MBSR-213 from Changjiang (19°28'19"N, 108°53'33"E), and Saccostrea MBSR-214 from Lingshui (18°24'19"N, 110°4'7"E). After morphological identification, adductor-muscle tissues were removed and preserved in 95% ethanol. Approximately 25–30 mg of muscle tissue was used for genomic DNA extraction with the TIANGEN Marine Animal DNA Kit (TIANGEN, China). DNA quality was evaluated via 1.0% agarose gel electrophoresis, and qualified samples were subsequently sent to Novogene in Beijing for high-throughput sequencing.
Illumina sequencing and sequence assembly
DNA libraries with insert fragments of about 300 bp were created using the NEB Next® Ultra™ DNA Library Prep Kit (NEB, USA) following the manufacturer’s instructions. Sequencing was performed on the Illumina NovaSeq 6000 platform, yielding 150 base pair paired-end reads. Quality control of the raw data was conducted using Fastp (Chen et al. 2018), with the adapters and low-quality reads removed. Subsequently, the clean data were imported into Geneious Prime 2025.0.2 (Kearse et al. 2012) to assemble the mitochondrial genomes, with “S. cuccullata” (KP967577) as the reference template.
Mitochondrial genome annotation and basic feature analysis
The mitochondrial protein-coding genes (PCGs) were predicted using MITOS WebServer (Bernt et al. 2013) and ORF Finder (Rombel et al. 2002) with the invertebrate genetic code table. The ribosomal RNA (rRNA) and transfer RNA (tRNA) genes were identified using WebServerand (Laslett and Canbäck 2008), respectively. The secondary structures of predicted tRNAs were visualized using (Darty et al. 2009). The nucleotide composition of the mitochondrial genome, including PCGs, tRNAs, and rRNA genes, was calculated using MEGA 11 (Tamura et al. 2021). Base skew values for a given strand were calculated as follows: AT skew = (A − T) / (A + T), GC skew = (G − C) / (G + C), where A, T, G, and C represented the counts of the four nucleotides. Codon usage in PCGs was analyzed using MEGA 11 with the invertebrate genetic code table. Additionally, the values of Ka, Ka and Ka/Ks ratios among the four species were calculated using DnaSP 6 (Rozas et al. 2017).
Phylogeny and OTU delimitation of the genus Saccostrea
To determine the systematic status of the four specimens, all Saccostrea mitochondrial genomes available on GenBank were included for phylogenetic analyses. Based on previous results (e.g. Cui et al. 2021), S. mordax was set as the root of the phylogenetic tree. The nucleotide sequences of 12 PCGs were codon-aligned and concatenated for subsequent analyses. The optimal partitioning scheme and corresponding evolutionary models were determined using PartitionFinder 2 (Lanfear et al. 2016). Bayesian inference (BI) was implemented using MrBayes 3.2.6 (Ronquist et al. 2012), running Markov chain Monte Carlo (MCMC) simulations for 10 million generations with sampling every 1,000 generations. The first 25% of samples were discarded as burn-in. Maximum-likelihood (ML) analysis was conducted using IQ - TREE 1.6.12 (Nguyen et al. 2014), allowing partitions to have different evolutionary rates and performing 10,000 ultrafast bootstrap replicates. The resulting phylogenetic trees were visualized using FigTree 1.4.5. The nucleotide sequences of 12 PCGs of Saccostrea from phylogenetic analysis was uploaded to the online ASAP (https://bioinfo.mnhn.fr/abi/public/asap/asapold.html) platform for OTU delimitation, with the K80 model selected and other parameters set to default (Puillandre et al. 2021). The ML tree was uploaded to the mPTP online server (https://mcmc-mptp.h-its.org/) to perform OTU delineation under default parameters (Kapli et al. 2017). Subsequently, genetic distances between different OTU groups were calculated using the K80 model in MEGA 11.
Results
Mitochondrial genome characteristics
The complete mitochondrial genomes of four Saccostrea specimens were successfully assembled, with sizes ranging from 16,280 bp (Saccostrea MBSR-214) to 16,289 bp (Saccostrea MBSR-212). All four mitogenomes exhibit identical gene organization, encoding 12 PCGs, 23 tRNAs, and 2 rRNAs on a single strand, with the distinctive feature of a fragmented 16S rRNA gene (Suppl. material 1: tables S1–S4). Comparative analyses revealed consistent nucleotide composition biases across genomic elements (Fig. 1). AT-skew values demonstrated thymine preference in whole genomes (−0.044 to −0.240), PCGs, and tRNAs, while rRNA genes showed adenine bias (0.011–0.021). All genomic components exhibited guanine preference in GC-skew (0.196–0.204). Secondary structure prediction confirmed that all tRNAs maintain canonical cloverleaf conformations, including the duplicated tRNA-Met, tRNA-Ser, and tRNA-Leu genes (Fig. 2). Initiation codon analysis showed that most PCGs employed ATG start codons, with exceptions including GTG-initiated ND3, ND5, and ND2 (except Saccostrea MBSR-214 ND2 which used ATG), and ATA-initiated CYTB. All PCGs terminated with complete stop codons (TAA in 18 genes; TAG in 30 genes). The four species displayed nearly identical codon usage patterns (Fig. 3), with UUU, UUA, UUG, AUU, AUG, GUU, UCU, GCU, and GGG being predominant codons and CGC being the rarest. Leucine represented the most abundant amino acid, while glutamate was the least frequent. Evolutionary analysis indicated limited nonsynonymous substitutions among the four species, with most variations being synonymous. The COX1 gene exhibited the strongest purifying selection (average Ka/Ks = 0.0026), while ND2 and ND6 showed relatively higher Ka/Ks ratios (Fig. 4).
Variations in base composition of the mitochondrial genomes of four Saccostrea specimens.
Inferred secondary structures of 23 tRNA genes in four mitochondrial genomes.
A. Relative Synonymous Codon Usage (RSCU); B. Frequency of four mitochondrial genomes.
A. Values of non-synonymous (Ka) and synonymous (Ks) substitution; B. Ka/Ks ratios of each PCG.
Phylogenetic relationships and OTU delimitation
Phylogenetic reconstruction based on 12 PCGs from 31 Saccostrea mitochondrial genomes yield concordant topologies through ML and BI analyses, consistently resolving five major clades within the genus (Fig. 5). This study employed two delimitation methods (ASAP and mPTP) and identified nine OTUs within the genus Saccostrea based on mitochondrial coding sequences. Among these, OTU1, OTU3, OTU6, OTU8, and OTU9 match previously described species (Li et al. 2021; Cui et al. 2021), while the remaining OTUs represent currently unrecognized lineages. Phylogenetically, the nine OTUs are distributed across five major clades. The uppermost clade contains OTU1 and OTU2, which encompass several misannotated Saccostrea mitogenomes from GenBank. The second clade consists of sister lineages S. kegaki (OTU3) and Saccostrea MBSR-214 (OTU4). The third clade includes Saccostrea MBSR-211–213, all grouped under OTU5. The fourth clade comprises S. malabonensis (OTU6) and OTU7, and the fifth clade corresponds to the S. mordax complex (OTU8 and OTU9). Notably, OTU4 and OTU5 show pronounced genetic divergence (>10%) from all other OTUs. Within OTU5, pairwise genetic distances are relatively low: 1.7% between S. MBSR-211 and S. MBSR-212, 1.6% between Saccostrea MBSR-211 and Saccostrea MBSR-213, and 1.9% between Saccostrea MBSR-212 and Saccostrea MBSR-213.
Phylogenetic relationships of Saccostrea and species delimitation schemes based on the concatenated nucleotide sequences of 12 mitochondrial protein-coding genes. The first number at each node is bootstrap proportion (BP) of maximum likelihood (ML) analyses, and the second number is Bayesian posterior probability (PP). Nodal with maximum statistical support (BP = 100; PP = 1) is marked with a solid red circle.
Discussion
The structural characterization of the four Saccostrea mitochondrial genomes obtained in this study aligned with those of other members of the genus. The absence of the ATP8 gene, a common occurrence in bivalves, was also observed here (Zhan et al. 2018; Zhou et al. 2024). Furthermore, the fragmentation of the large subunit 16S rRNA into two segments is a distinctive feature of oysters (Xiao et al. 2015; Milbury et al. 2010). The intergenic region between COX1 and tRNA-Thr exhibited a high AT content, suggesting its putative role as a control region for replication and transcription. Evolutionary analyses revealed strong purifying selection pressures acting on all PCGs, with Ka/Ks ratios consistently below 1.0. This suggests that natural selection favours the preservation of the original gene state, thereby inhibiting the accumulation of mutations (Hurst 2002).
Salvi et al. (2014) first proposed a three-subfamily phylogenetic framework for the family Ostreidae, including Crassostreinae, Saccostreinae, and Ostreinae. Subsequent studies further refined this framework and supported the currently accepted phylogenetic classification of Ostreidae, which includes Crassostreinae (comprising Crassostrea Sacco, 1897; Talonostrea Li & Qi, 1994; and Magallana), Saccostreinae (comprising Saccostrea Dollfus & Dautzenberg, 1920), Striostreinae (comprising Striostrea Vialov, 1936), and Ostreinae (including the remaining genera) (Raith et al. 2016; Salvi and Mariottini 2016). A characteristic feature of Saccostrea is the presence of small chomata along the inner edge of the shell (Lu et al. 2024). Within the genus Saccostrea, morphological variation enables further species-level discrimination. The diagnostic criterion for S. mordax involves its distinctive wavy shell margins that demonstrate continuous extension into the ligament area, a morphological signature first systematically described by Amaral and Simone (2016). This characteristic feature serves as a reliable morphological marker for distinguishing S. mordax from other congeneric species within the genus.
At the molecular level, the two representatives of S. mordax are robustly segregated from other Saccostrea lineages in the phylogeny, congruent with morphological assessments of Saccostrea. Conversely, non-S. mordax taxa exhibit pronounced taxonomic incongruence. The pronounced plasticity of oyster shells may mislead non-specialists, resulting in erroneous species assignments and the deposition of mislabelled sequences in GenBank (Cui et al. 2021; Crocetta et al. 2015). For instance, when studying small alien oysters in the Mediterranean, Crocetta et al. (2015) reported that 16S and COI sequences assigned to the genus Dendostrea in GenBank did not form a monophyletic clade, with many sequences originating from misidentified specimens. Sequence annotations for the genus Dendostrea and related taxa in GenBank are mostly based on shell morphological characteristics and geographic distribution inferences, rather than rigorous integrated molecular and morphological identification, leading to confusion in species delimitation (Oliver et al. 2025; Delcour et al. 2025). Similarly, Sigwart et al. (2021), while describing the new species Crassostrea (Magallana) saidii, confirmed that approximately 5% of COI sequences in the Magallana clade were either misidentified or lacked reliable species annotations. Moreover, Hu et al. (2019), in their taxonomic revision of the Ostrea stentina species complex, demonstrated that previous sequence annotations of several closely related oysters in GenBank had obscured true species boundaries, underscoring that such classification biases can only be corrected by integrating morphological traits with multilocus molecular data. Historically, oyster systematics relied exclusively on conchological characters. The advent of DNA barcoding subsequently introduced 16S rRNA and COI in contemporary barcoding approaches. Nevertheless, persistent incongruence among morphological, 16S rRNA, and COI datasets creates substantial challenges for achieving unified species delineation. This discordance suggests two opposing systematic issues: potential over splitting leading to synonymy proliferation, versus undetected cryptic diversity masked under single operational taxonomic units (OTUs). Mitogenomes are therefore expected to serve as a powerful tool for data integration across morphological, COI, and 16S datasets.
Notably, within OTU5, pairwise genetic distances range from 1.6% to 1.9%, suggesting that these three samples may represent incipient subspecies or independently evolving specimens (Torstrom et al. 2014; Chen et al. 2024). Indeed, more sensitive delimitation approaches could potentially split OTU5 into three distinct OTUs (data not shown). However, in the absence of robust biological evidence—such as genomic data, geographically distributed populations, or hybridization experiments—we adopt a conservative taxonomic stance and currently treat them as a single OTU. Future studies incorporating broader sampling and multidimensional molecular datasets will be essential to clarify the true evolutionary relationships among these three individuals.
Conclusion
This study elucidates the complete mitochondrial genomes of four species (Saccostrea MBSR-211–214) from Hainan Island, revealing conserved structural features consistent with congeneric organisms. By integrating phylogenetic reconstruction, and OTU delimitation analysis, it confirms their taxonomic status as distinct specimens within Saccostrea. A larger sample set and higher-dimensional molecular data will be essential to clarify the taxonomic status of these lineages. Our findings again underscore the current unreliability of Saccostrea nomenclature in public databases such as GenBank. Moving forward, it may be feasible to establish a standardized molecular reference database for oysters through the integrated use of mitochondrial genomes and nuclear gene markers. As biodiversity reassessment progress, enhanced in nomenclature is imperative to avoid taxonomic ambiguities.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Amaral VS, Simone LRL (2016) Comparative anatomy of five species of Saccostrea Dollfus & Dautzenberg, 1920 (Bivalvia: Ostreidae) from the Pacific Ocean. The Nautilus 130(2): 53–71. 10.5281/zenodo.15938620 · doi ↗
- 2Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution 69(2): 313–319. 10.1016/j.ympev.2012.08.02322982435 · doi ↗ · pubmed ↗
- 3Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17): i 884–i 890. 10.1093/bioinformatics/bty 560PMC 612928130423086 · doi ↗ · pubmed ↗
- 4Chen Y, Li C, Lu R, Wang H (2024) Morphological and molecular analysis identified a subspecies of Crassostrea ariakensis (Fujita, 1913) along the coast of Asia. Genes 15(5): 644. 10.3390/genes 15050644 PMC 1112121238790273 · doi ↗ · pubmed ↗
- 5Crocetta F, Mariottini P, Salvi D, Oliverio M (2015) Does Gen Bank provide a reliable DNA barcode reference toidentify small alien oysters invading the Mediterranean Sea? Journal of the Marine Biological Association of the United Kingdom 95(1): 111–122. 10.1017/S 0025315414001027 · doi ↗
- 6Cui Z, Hu L, Li C, Zhang Z, Guo X, Wang H (2021) Identification of Saccostrea mordax and a new species Saccostrea mordoides sp. nov. (Bivalvia: Ostreidae) from China. Journal of Shellfish Research 40(1): 63–75. 10.2983/035.040.0107 · doi ↗
- 7Darty K, Denise A, Ponty Y (2009) VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics: 25(15): 1974–1975. 10.1093/bioinformatics/btp 250PMC 271233119398448 · doi ↗ · pubmed ↗
- 8Delcour N, Garzia M, Oliver PG, Berrilli E, Toso A, Bariche M, Albano PG, Mariottini P, Salvi D (2025) High genetic diversity and lack of structure underlie the invasion history of the non-indigenous oyster Dendostrea cf. crenulifera (Mollusca, Ostreida, Ostreidae) spreading in the eastern Mediterranean Sea. Neo Biota 101: 277–302. 10.3897/neobiota.101.154917 · doi ↗