Emerging role of nucleic acid aptamers in sepsis-induced coagulopathy: Future perspectives in diagnostics and therapeutics
Maeva Martin, Marine Tschirhart, Cyril Auger, Florence Toti, Julie Helms, Laurence Choulier

TL;DR
This review explores how nucleic acid aptamers could be used to diagnose and treat sepsis-induced coagulopathy, offering new hope for patients.
Contribution
The paper highlights the novel potential of nucleic acid aptamers in targeting multiple aspects of sepsis-induced coagulopathy.
Findings
Aptamers can target endothelial dysfunction to protect vascular integrity and reduce inflammation.
They can modulate coagulation pathways by blocking platelet aggregation and coagulation factors.
Aptamers may restore fibrinolytic function and inhibit the complement system in sepsis.
Abstract
Sepsis-induced coagulopathy is a severe complication of sepsis and septic shock, contributing significantly to organ dysfunction and increased mortality. Currently, there is no effective treatment for coagulopathy, highlighting a critical need for new therapeutic options to improve patient outcomes. This review explores the potential use of nucleic acid aptamers for septic-induced coagulopathy. Aptamers are short single-stranded oligonucleotides, known for their high affinity and specificity towards targets. They offer several advantages over antibodies, including smaller size, synthetic production, lower immunogenicity, and greater flexibility in targeting a wide range of molecules and cells. Due to these properties, aptamers have emerged as promising tools for the diagnosis and treatment of various diseases, with several currently ongoing clinical trials and others already available…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
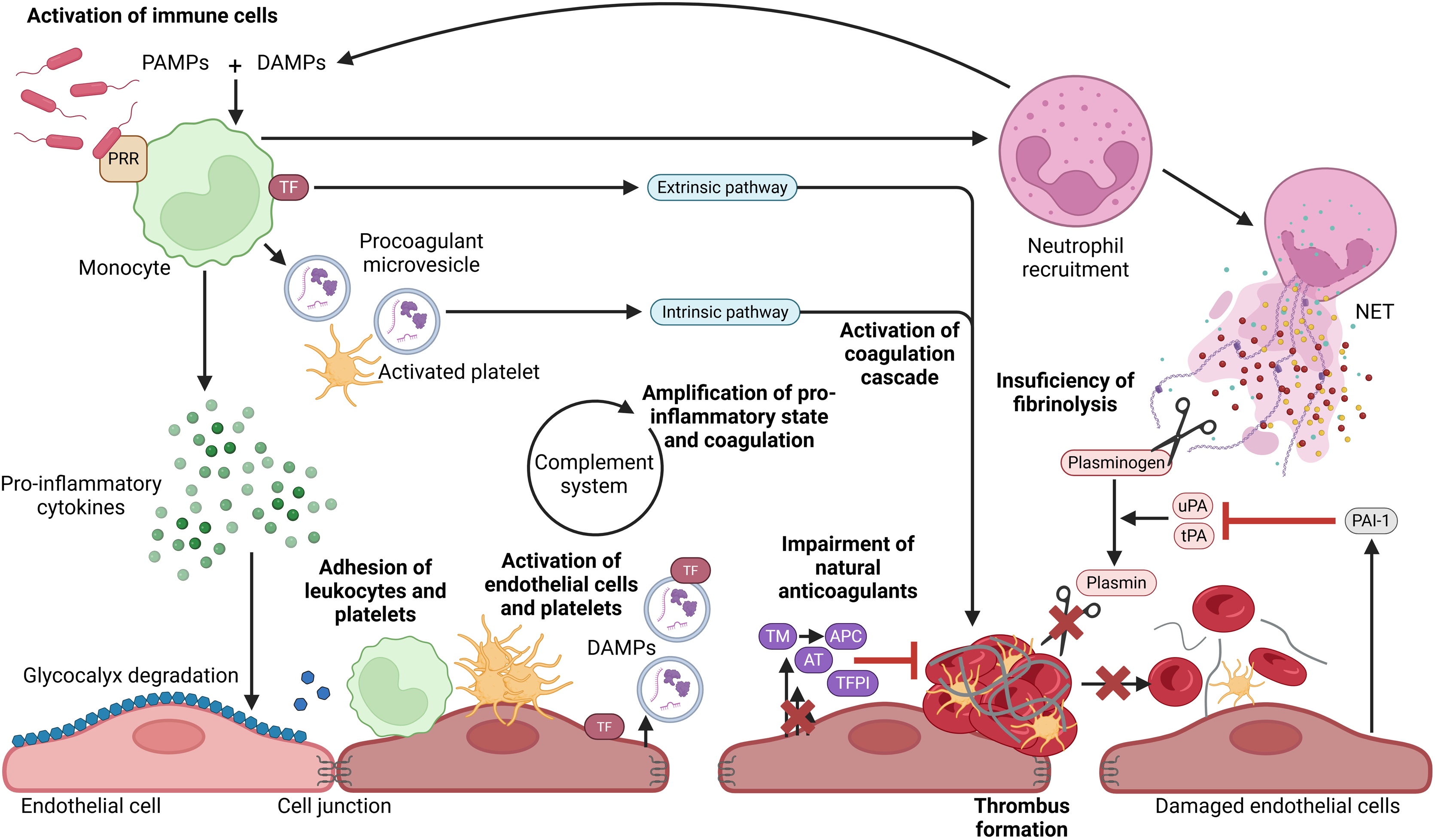 Figure 1
Figure 1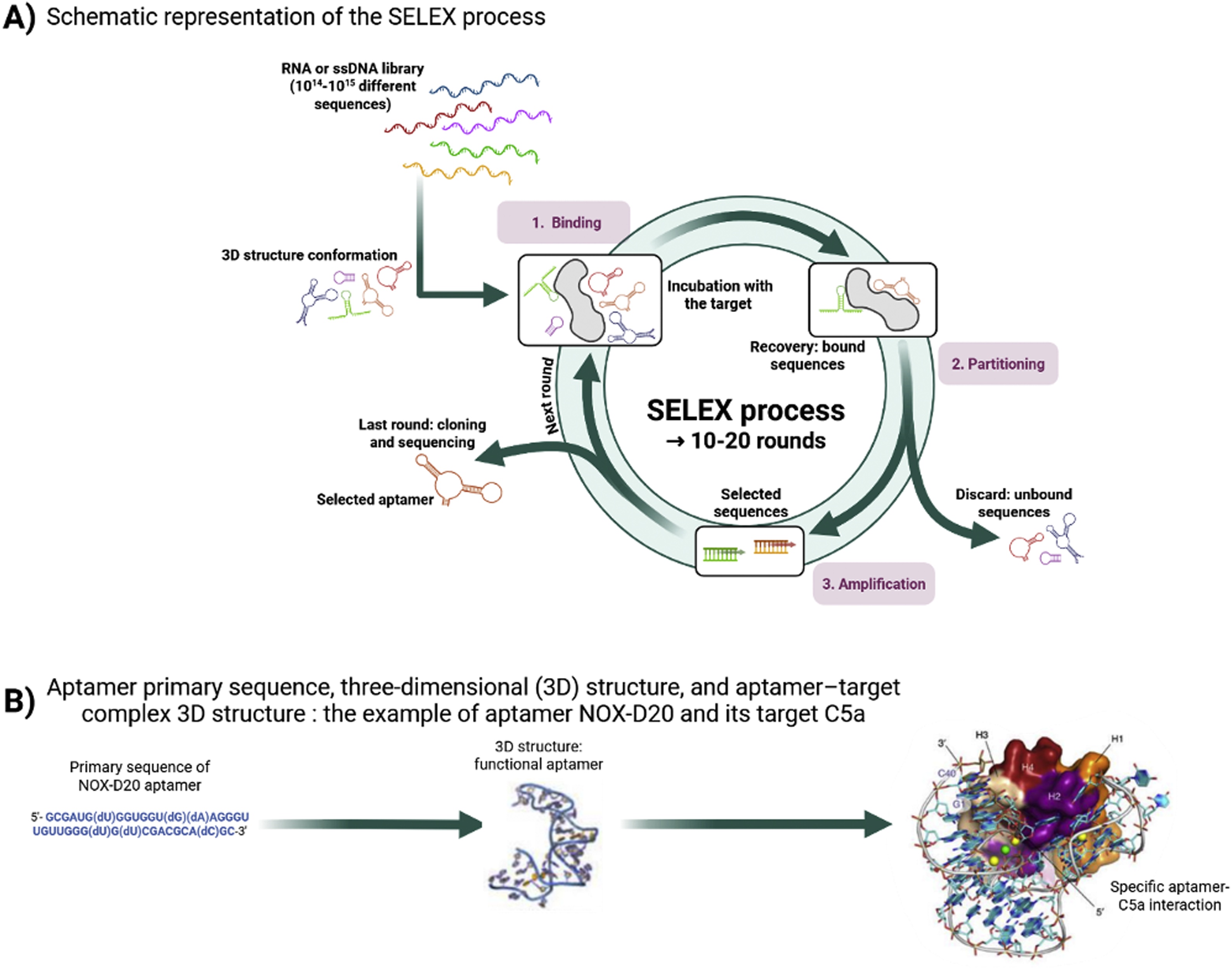 Figure 2
Figure 2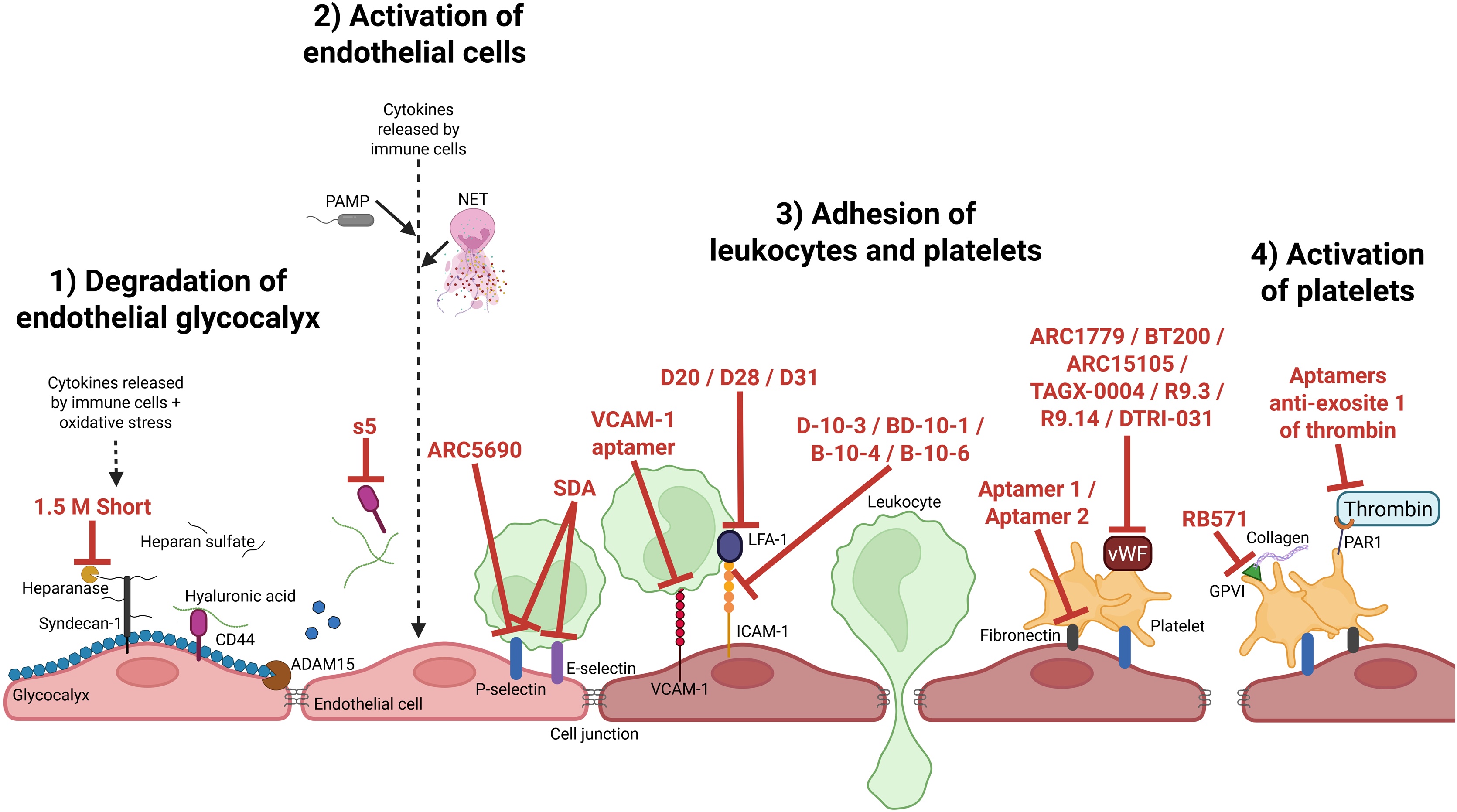 Figure 3
Figure 3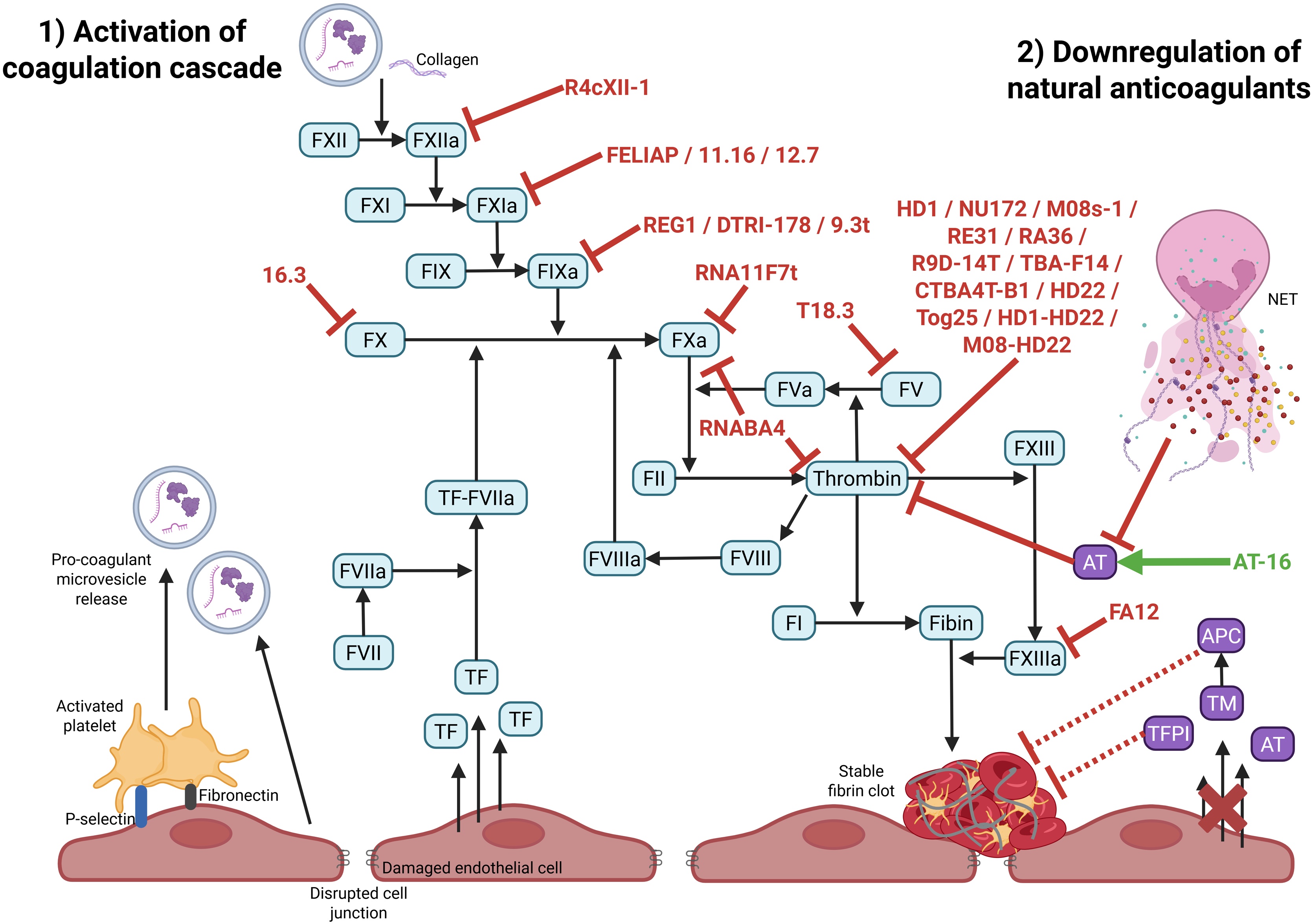 Figure 4
Figure 4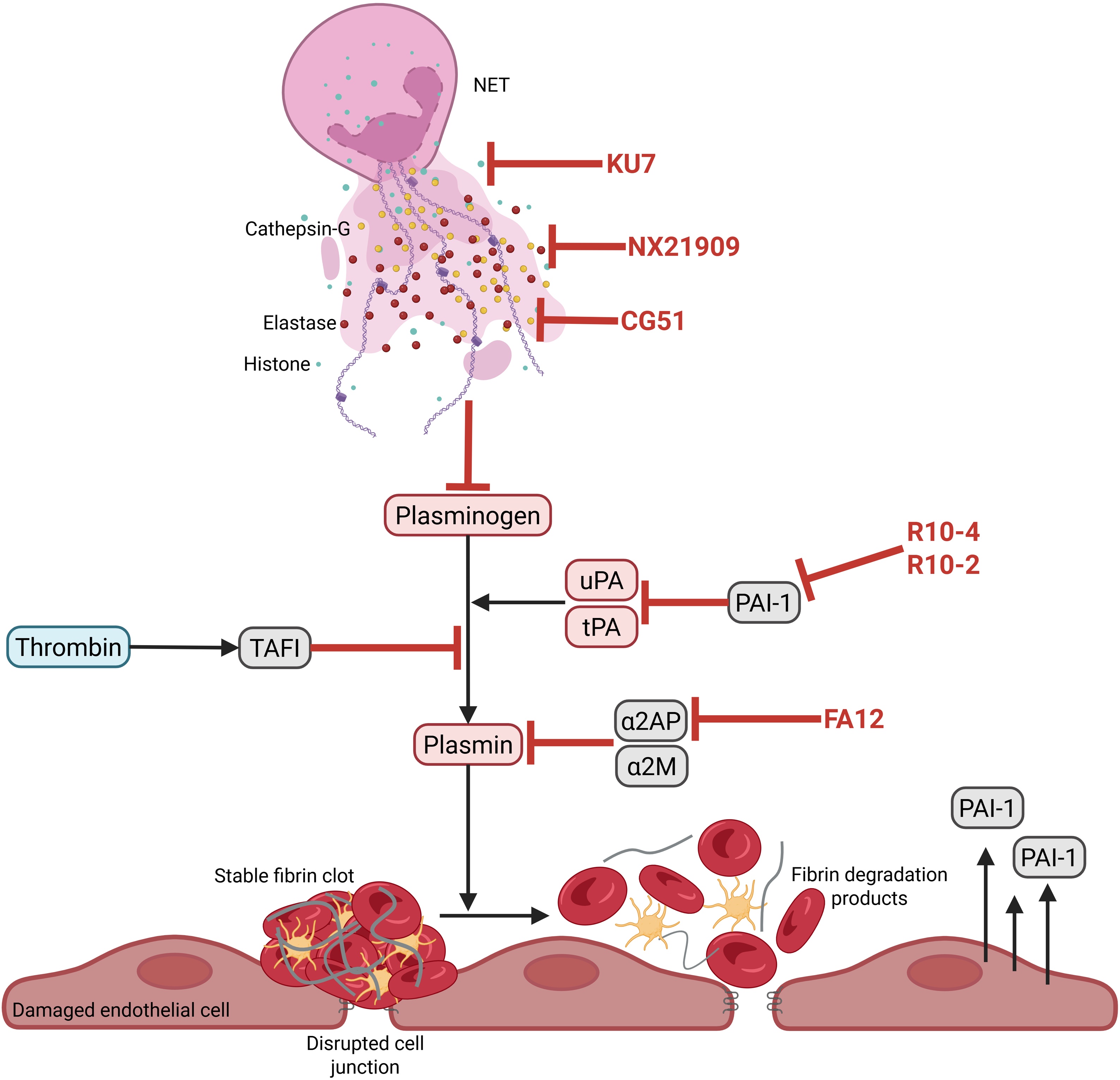 Figure 5
Figure 5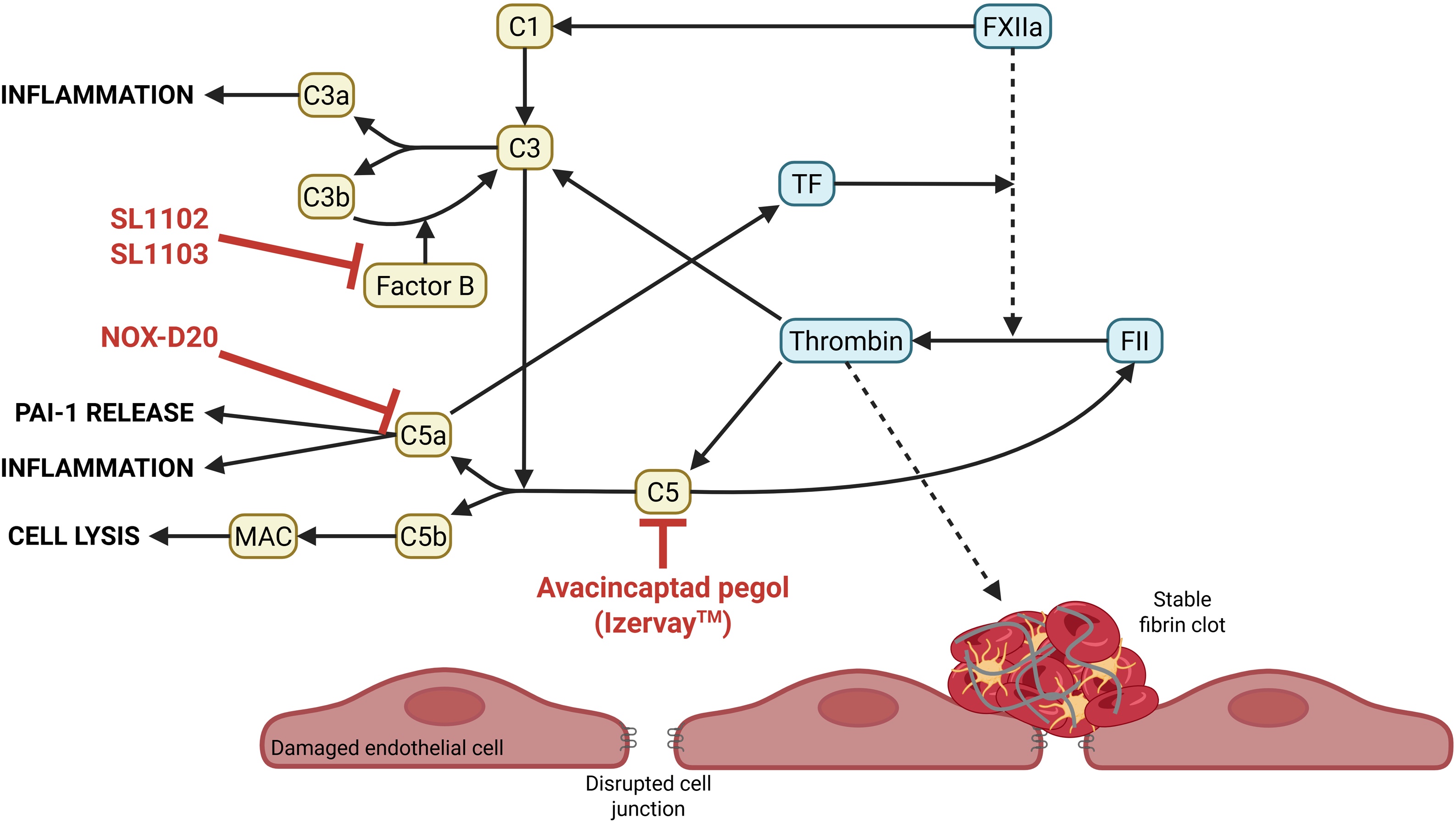 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced biosensing and bioanalysis techniques · Neutrophil, Myeloperoxidase and Oxidative Mechanisms · Cell Adhesion Molecules Research
Introduction
Sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection, and may progress to septic shock, a severe form characterized by circulatory and metabolic abnormalities [1]. In 2020, the WHO identified sepsis as a global health priority, with 49 million cases and 11 million deaths reported in 2017 [2].
Sepsis can be triggered by various pathogens (bacteria, viruses, fungi, parasites), which activate local pro-inflammatory and procoagulant responses. The interaction between immunity and coagulation, termed immunothrombosis, helps contain infection through innate immune cell recruitment [3,4]. However, in sepsis, this process becomes dysregulated, promoting endothelial damage and excessive coagulation. Hyperinflammation is further amplified by complement activation and coagulation imbalance [5]. This may lead to coagulopathy and disseminated intravascular coagulation (DIC), marked by widespread microthrombosis and organ dysfunction, with mortality rates up to 65% in septic DIC [6]. Sepsis-induced DIC is also characterized by impaired fibrinolysis, partly due to neutrophil extracellular traps (NETs)-induced plasminogen degradation and endothelial release of plasmin inhibitors.
There is currently no specific treatment for sepsis-induced DIC. Management is limited to infection control and organ support, and no validated markers exist for risk stratification. In sepsis-induced DIC, nucleic-acid aptamers represent promising tools for both improving early diagnosis and enabling targeted therapy, which could potentially minimize off-targets and adverse effects of potential treatments.
This review highlights potential aptamer-based strategies for sepsis-induced coagulopathy, with focus on endothelial dysfunction, coagulation imbalance, hypofibrinolysis, and diagnosis.
Pathophysiology of septic DIC
Since sepsis results from a dysregulated host response to infection, the first step involves immune system activation via interactions between pathogen-associated molecular patterns (PAMPs) and pattern recognition receptors (PRRs). Monocytes and neutrophils then release pro-inflammatory cytokines and procoagulant microvesicles. This hyperinflammatory state contributes to endothelial glycocalyx degradation, increasing permeability and exposing the endothelial surface to procoagulant stimuli. Damaged endothelial cells release damage-associated molecular patterns (DAMPs), amplifying systemic inflammation.
Inflammatory cytokines also upregulate tissue factor (TF) on immune and endothelial cells, initiating the extrinsic coagulation pathway. Endothelial activation further promotes immune cell adhesion and secretion of von Willebrand factor (vWF), facilitating platelet aggregation and thrombus formation. This process, known as immunothrombosis, helps trap and neutralize pathogens within the thrombus [7].
In septic DIC, immunothrombosis becomes dysregulated, with excessive coagulation, reduced anticoagulants (antithrombin, thrombomodulin), insufficient TF pathway inhibition, and defective fibrinolysis [[8], [138]]. Procoagulant microvesicles and increased TF expression drive clot formation, while fibrinolysis is impaired by reduced plasmin generation—due to endothelial-derived plasminogen activator inhibitors and NET-mediated plasminogen degradation [9]. NETs are DNA-based structures released during NETosis, a specific form of neutrophil cell death. Complement activation further amplifies inflammation and coagulation by releasing anaphylatoxins and enhancing TF expression, in part via a self-amplification of the complement system [8]. These mechanisms are summarized in Fig. 1.Fig. 1Physiopathology of septic DIC. Activated immune cells release cytokines which leads to the degradation of the endothelial glycocalyx, contributing to endothelial cell activation and leukocytes and platelet adhesion on endothelium. Coagulation cascade, which is activated by phosphatidylserine exposed on microvesicles, activated platelets and TF expression on immune and endothelial cells, results in microthrombus formation. Furthermore, pro-inflammatory state and damaged endothelial cells are associated to downregulation of endogenous anticoagulants and to decrease of fibrinolysis. Additionally, complement system interacts with coagulation cascade to activate each other and amplify pro-inflammatory state by its anaphylatoxin release. PAMP: pathogen associated molecular pattern, DAMP: damage associated molecular pattern, PRR: pattern recognition receptor, TF: tissue factor, NET: neutrophil extracellular trap, AT: antithrombin, TM: thrombomodulin, APC: activated protein C, TFPI: tissue factor pathway inhibitor, uPA: urokinase plasminogen activator, tPA: tissue-type plasminogen activator, PAI-1: plasminogen activator inhibitor type-1.Fig. 1
Considering this complexity, innovative tools are needed to both better characterize and modulate these pathways. Among them, nucleic-acid aptamers have emerged as promising candidates due to their ability to specifically bind and modulate diverse molecular targets implicated in sepsis-induced coagulopathy.
Nucleic acid aptamers
Naturally occurring nucleic acid aptamers, known as riboswitches, are RNA elements that regulate gene expression by binding specific metabolites or ions within cells. They illustrate the intrinsic ability of nucleic acids to form complex three-dimensional structures capable of high-affinity molecular recognition. However, in the context of this review, the focus will be on synthetic aptamers. They are short single-stranded DNA or RNA sequences that act as high-affinity and high-specificity ligands for a wide variety of targets. These targets range in nature and size—from small molecules (metabolites, toxins, drugs) to macromolecules (proteins, lipids, nucleic acids), pathogens (viruses, bacteria), organelles, and even whole cells [10]. This versatility distinguishes aptamers from antibodies, especially since they can also target non-immunogenic or toxic molecules [11].
Aptamers are identified through an in vitro selection method called SELEX (Systematic Evolution of Ligands by EXponential enrichment) [12,13]. Starting from a vast random library (∼10^15^ oligonucleotide sequences), SELEX combines repeated cycles of target binding, separation of bound from unbound sequences, and amplification to enrich for high-affinity ligands (Fig. 2). Several SELEX variants have been developed to enhance aptamer performance under physiological or complex conditions, thereby improving affinity and specificity.Fig. 2From SELEX to aptamer–target recognition. (A) Schematic representation of the SELEX (Systematic Evolution of Ligands by Exponential Enrichment) process leading to the selection of high-affinity nucleic acid ligands. Briefly, the nucleic acid library is incubated with the target of interest. During the partitioning step, bound and unbound fractions are separated. The bound fraction is amplified to obtain an enriched pool for next round of selection. This round is repeated between 10 and 20 times to enrich the sequences that specifically bind the target. At the last round, nucleic acid sequences, named aptamers, are cloned and sequenced. (B) The example of NOX-D20 aptamer and its target C5a: aptamer primary sequence, three-dimensional (3D) structure, and aptamer-target complex 3D structure [137].Fig. 2
Compared to antibodies, aptamers offer major advantages in terms of chemical synthesis, thermal stability, and flexibility. They have distinctive pharmacokinetic and structural properties that confer them both advantages and limitations, which can be modulated through diverse chemical and structural modifications to enhance their stability and bioavailability for therapeutic use (Table 1).Table 1. Characteristics of aptamers versus antibodies. Improvement strategies are shown as footnotes.Table 1. PropertyAptamerAntibodyChemical propertyNucleotidesAmino acidsSpecificity and affinityHighHighTime needed for selectionSeveral weeksSeveral monthsCost of selection≈ 8,000 for mouse monoclonal antibody≈$ 20,000 for rabbit monoclonal antibodySynthesis and manufactureChemical synthesisHigh scale productionChemical modificationsProduced in animal or by recombinant methodsModificationEasy and controllableLimited and uncontrollableStorage and stabilityStable under different conditions Can recover their native structure after denaturationRequires special conditions for storage and handlingEnzymatic degradationNuclease degradation (mainly for RNA)^1^Little degradationBatch-to-batch variationsLittle or noDifficult to avoidSize5−30 kDa allows rapid diffusion and increased tissue penetration^2^Whole size of antibody is 150 kDaTarget rangeLarge: small molecule, protein, cell…Limited to antigenic targetsIn vivo complicationsNo evidence of immunogenicity or toxicityImmune response to (modified) aptamers is theoretically possible, but has not been described to date.May lead immune responseClinical applicationStill immatureMatureImprovement strategies: ^1^Chemical modifications: Sugar modifications (e.g., 2’-fluoro, amino, or methyl substitutions), Backbone modifications, such as replacing phosphodiester linkages with triazole groups (via click chemistry) or using phosphonothioate bonds, Base modifications (e.g., C5 of pyrimidines or N7 of purines), Circularization of aptamers, Spiegelmers: L-enantiomeric versions of natural D-aptamers. ^2^.To limit rapid renal clearance: Terminal modifications at the 3’ or 5’ ends with carriers (e.g., with biotin, cholesterol, or polyethylene glycol [PEG], poly(lactic-co-glycolic acid) [PLGA], or proteins), Multimerization.
Another distinctive feature of aptamers is the possibility to reverse their effect using specific antidotes—complementary oligonucleotide strands that competitively bind to the aptamer and block its interaction with the target [14]. This controllability is particularly attractive for therapeutic use. Aptamers also show promise in pathogen detection, with high sensitivity and specificity [[15], [16], [17]], and in anti-infective strategies, where they could help overcome antimicrobial resistance [18].
In the following sections, we will explore how aptamers may be harnessed to target the key pathological features of sepsis-induced coagulopathy (Table 2) — namely endothelial dysfunction, coagulation imbalance and hypofibrinolysis, as well as used as diagnostic and prognostic tools (Table 3).Table 2. Summary table of aptamers with potential therapeutic effects in sepsis-induced coagulopathy.Table 2. NameTypeSequence 5’ → 3’TargetLevel of studyPossible effect on septic DICReference1.5M ShortDNAACTTTTGAATGTGGCAACAAATTGACAGGheparanaseIn vitroPreserve endothelial glycocalyx(20)s5DNAACCGGGCGTACACCGTCGCGGCACATGTCTGAATGCGTTTAGTCTCTGTGCD44In vitroMaintain low vascular permeability(21)D-10-3DNAAGAGACCCTGACTGCGAACTCCCACTTTACCTCTTCCCTCAACTTCCCCTTCCTTTCTGACCCTGGACACGGTGGCTTCTTICAM-1In vitroLimit leukocyte adhesion and transmigration(26)BD-10-1DNAAGAGACCCTGACTGCGAACATATACAGACGGTAGCGACTGGAAGAGAGCCTTAACCACACATCTGGACACGGTGGCTTCTTB-10-4DNAAGAGACCCTGACTGCGAACACACATGGAACGCAACACCCTGAAACTATGGACTGACGAGACCATGGACACGGTGGCTTCTTB-10-6DNAAGAGACCCTGACTGCGAACCAACCACAGCTTAATATCCTCATATCCGCGACCGCTCGCGAGCATGGACACGGTGGCTTCTTVCAM-1 aptamerRNAAGGGAAUCUUGCCUAGGGAGGGAGUAGCGAAAGGGCUCAVCAM-1In vivoLimit leukocyte adhesion and transmigration(27)D20RNAUGCGCGUCCCAUGGGGUAUAGAGGGGUCGAAGUGGACGCGLFA-1In vitroReduce cell adhesion to ICAM-1(28)D28RNAUCCUCGCUCAUACAACGAGGGGUCGUGUAGGGAUGUAUGGGD31RNAGGGCGCUAAGUCCUCGCUCACAAGGUGCAAUGCAAUAUGUGAGUGCGCCGCCCUUUCUCUCGCGCGACUCGGACCUACARC5690RNA40 kDa PEG – CUCGCAGACAACCGGAUGAAAUCCGACCGGAG - idTP-selectinIn vivoLimit leukocyte adhesion and rolling(29)SDADNAGCCTGTTGTGAGCCTCCTAACGATTTGGATTTGGGGTGGAGGGTATGGTTTGTGCTGGCGTTCTCATTTCCCATGCTTATTCTTGTCTCCCE- and P-selectinIn vitroLimit leukocyte adhesion and rolling(30)Aptamer 1DNACATGCGCCTTCCCCCTGTGGTTGGTGTCAGTCGGCCTGTGTGATTAGTGGAATGTGCGATGGGTCTCCCAGCGGGATTGGFibronectinIn vitroLimit adhesion of platelets to the sub-endothelium(32)ARC1779DNA/RNA20 kDa PEG – GCGUGCAGUGCCUUCGGCCGTGCGGTGCCUCCGUCACGC – idT2-O-Methyl nucleotidevWFPhase IIReduce platelet aggregationNCT00726544NCT00632242NCT00694785NCT00742612NCT00507338NCT00432770BT200RNA40 kDa – GCCAGGGACCUAAGACACAUGUCCCUGGC – idT2-O-Methyl nucleotidesPhase IINCT04677803 NCT04103034ARC15105RNA40 kDa PEG – NH -GGGACCUAAGACACAUGUCCC-idT2-O-Methyl nucleotidesIn vivo(41)TAGX-0004DNACGCGGCCGADsGAGCACCGAAGGTCTCAACGDsTGGAGGCCGCGCGCGT’AGCGIn vitro(42)R9.3DNAATCGCGCTCTCCTGCTTAAGCAGCTATCAAATAGCCCACTin vitro(43)R9.14DNATGGACGAACTGCCCTCAGCTACTTTCATGTTGCTGACGCADTRI-031RNAACGAACUGCCCUCGAUCGACGCACAGACGUUUUUU2-O-Methyl nucleotides2’-Fluoro nucleotides**In vivo(44)RB571RNA29-nucleotide modified RNA however no more information is givenGPVIIn vitroInhibits collagen-related peptide mediated platelet aggregation(45)R4cXII-1RNAGGGAGGACGAUGCGGCCAAAUCUCGGCUGCCAGCAGGUCACGAGUCGCAAGAUAACAGACGACUCGCUGAGGAUCCGAGA2’-Fluoro nucleotidesFXIIIn vitroProlong blood clot formation(48)FELIAPDNAAACCTATCGGACTATTGTTAGTGATTTTTATAGTGTFXIaIn vitroExtend clotting times and inhibit thrombin generation(49)11.16RNACCAGUCCGCAUCCAUCAUCCCCCUCCCCCIn vitro(50)12.7RNAUAACGCCACGCUCGACAACGCGUCGAGUGUCCUCCGCCCCREG1: RB006 (drug) / RB007 (antidote)RNARB006: 40 kDa PEG – NH – GUGGACU AUACCGCGUAAUGCUGCCUCCAC – idTRB007: CGCGGUAUAGUCCCCAU2-O-Methyl nucleotides2’-Fluoro nucleotidesFIXaPhase IIIReduce thrombin generationNCT01848106NCT00715455NCT00932100NCT00113997NCT01872572DTRI-178 (RB006)RNA40 kDa PEG – NH – GUGGACUAUA CCGCGUAAUGCUGCCUCCAC – idT2-O-Methyl nucleotides2’-Fluoro nucleotides**In vivo(54)9.3tRNAAUGGGGACUAUACCGCGUAAUGCUGCCUCCCCAU - idTIn vitro(130)16.3RNAUACAGAGGAGUACAAGUAGCAUGAUCCCUCGUGUAAAFXIn vitroBlock the initiation of coagulation(55)RNA11F7tRNAGAGAGCCCCAGCGAGAUAAUACUUGGCCCCGCUCUUFXaEx vivoInhibit thrombin formation(57)RNABA4RNAGGUCGAUCACACAGUUCAAACGUAAUAAGCCAAUGUACGAGGCAGACGACAAAGCCCCAGCGAGAUAAUACUUGGCCCCGCUFXA + FIIaIn vitroReduce clot formation(58)T18.3RNAGGACUUGGAUAACCUCACCGCAAUGGCGGCUUGUCAGACGACUCGCUGAGGAUCCGAGFVIn vitroReduce clot formation(56)HD1DNAGGTTGGTGTGGTTGGFIIa (Thrombin)In vivoInhibit clot formation(61)NU172DNACGCCTAGGTTGGGTAGGGTGGTGGCGPhase IINCT00808964M08s-1DNAAGGTCAGATGATGGGGATGGGGGGTTGGAGGAATGGATGACCTIn vivo(14)RE31DNACACTGGTAGGTTGGTGTGGTTGGGGCCAGTGIn vitro(131)RA36DNAGGTTGGTGTGGTTGGTGGTTGGTGTGGTTGGIn vitro(132)R9D-14TRNAGGCGGUCGAUCACACAGUUCAAACGUAAUAAGCCAAUGUCGAGGCAGACGACUCGCC2’-Fluoro nucleotides**In vitro(133)TBA-F14DNAGGTTGGTGTGGTTGG8-trifluoromethyl-2′-deoxyguanosineIn vitro(134)CTBA4T-B1DNATAGGGGGCGCGAACATACGCGGTTGGTGTGGTTGGCTGACTCGTATCTCGAGTCAIn vitro(135)HD22DNAAGTCCGTGGTAGGGCAGGTT GGGGTGACTEx vivo(64)Tog25RNAGAACAAAGCUGAAGUACUUACCCAAGAUCAUCCCGAACGAIn vitro(65)HD1-HD22DNAGGTTGGTGTGGTTGGAAAAAAAAAAAAAAAAGTCCGTGGTAGGGCAGGTTGGGGTGACTIn vitro(66)M08s-1-HD22DNAAGGTCAGATGATGGGGATGGGGGGTTGGAGGAATGGATGACCTTTTTTTTTTTTTTTTAGTCCGTGGTAGGGCAGGTTGGGGTGACTIn vivo(14)FA12DNAAAGCAGTGGTAAGTAGGTTGATCATCCTCTTTAAGTCATCACTTTAGTTTCTCCATCTACATCTCTTCGAGCAATCCACACFXIIIaIn vitroReduce clot rigidity and can allow fibrinolysis(76)AT-16RNANot referencedAntithrombinIn vitroEnhance endogenous anticoagulant properties of antithrombin(70)R10-4RNACCAGGCGUCUCACUCGUUACGCUAUCGUUGCGUACUUCUGPAI-1In vitroinhibit PAI-1's antiproteolytic activity against tPA(74)R10-2RNACACACGAGGCAAGUGGCCUGCAUAACGUAGGCGUCGAGUAKU7RNAGGGAGGACGAUGCGGACUGGUGAAGGGAGGUACUGCAGACGACUCGCCCGAHistones H3 and H4In vivoInhibit histone-induced platelet aggregation(80)NX21909DNAAGGACGATGCGGCACGGTAGTGCTACCAGATGGTTATGTTAC + Splint oligo: valyl phosphonate - GGTGCCGCATCGTCCTNeutrophil elastaseIn vivoReduce the recruitment and influx of neutrophils(81)CG51DNACAACGTGTGATATGTGGGTATACGCTTGGGTGTTACGCTGAGCACAGAGGGTATTCGTGTCathepsin GIn vitroModulate proteolytic activity of cathepsin G(82)SL1102DNACGC-2NEdU-GAGAA-2NEdU-AGAAG-2NEdU-AGGAG-2NEdU-A-2NEdU-GC-2NEdU-2NEdU-GCG2-O-Methyl nucleotidesFactor BIn vitroRegulate the alternative pathway’s activity(86)SL1103DNAGC-2NEdU-GAGAA-2NEdU-AGAAG-2NEdU-AGGAG-2NEdU-A-2NEdU-GC-2NEdU-2NEdU-GC2-O-Methyl nucleotidesIzervay™RNA43 kDa PEG -CGCCGCGGUCUCAGG CGCUGAGUCUGAGUUUACCUGCG-idT2-O-Methyl nucleotides2’-Fluoro nucleotidesC5ApprovedPrevent C5 cleavage and decrease membrane attack complex formationNCT04435366NOX-D20RNA40 kDa PEG – GCGAUGUGGUGGU GAAGGGUUGUUGGGUGUCGACGCACGC DeoxyribonucleotidesC5aIn vivoAttenuate inflammation and organ damage(94)idT: inverted T nucleotides, NH: hexylamine linker, T’: biotin-dT, Ds: 7-(2-thienyl)imidazo[4,5-b]pyridine, 2NEdU: 5-[N-(2-naphthylethylcarboxyamide)-2′-deoxyuridine replace T nucleotides.Table 3. Summary table of aptamers with potential diagnostic applications for sepsis-induced coagulopathy.Table 3. NameTypeTargetDiagnosis / Prognosis toolsLimit of detection (LOD) or Affinity (K_D_)ReferenceNOX-S93RNASphingosine-1-phosphateLevel inversely related to endothelial glycocalyx degradationK_d_ = 4.3 nM(104)11-1.41RNAAngiopoietin 2Level related to increase of vascular permeabilityK_d_ = 2.2 nM(106)M55DNAThrombospodin-1Level related to endothelial dysfunctionLOD = 6.96 fM(110)Anti-kinin B1 receptor aptamerRNAKinin B1 receptorLevel related to hypotension and increased vascular permeabilityAffinity not determined(113)Anti-human neutrophil elastase aptamerDNANeutrophil elastaseLevel related to endothelial damage and coagulation cascade activationLOD = 20 fM(114)Anti-PCT aptamerDNAPCTLevel related to bacterial infectionsLOD = 0.01 ng/mL(97)CD63-1DNACD63Levels related to platelets in an active stateK_d_ = 38.71 nM(99)AT-1DNACD31K_d_ = 2.28 nM(100)iodo-FADNAFibrinogenLevel related to coagulation disorderK_d_ = 9.75 μM(136)HS02-88DNAAPCLevel inversely related to prothrombotic stateK_d_ = 0.43 nM(117)BAX499 / ARC19499RNATFPILevel inversely related to procoagulant effectsK_d_ = 2.8 nM(119,120)Aptamer 3218RNAtPACirculate tPA or uPA levels related to mortalityK_d_ ≈ nM(122)upanap-126RNAuPAAffinity not determined(123)WT-15DNAPAI-1Level related to thrombotic disordersK_d_ = 177 pM(125)If referenced, the dissociation constant is given for aptamers that are not yet developed as sensor tools.
Targeting the endothelial permeability and dysfunction
Inflammatory stimuli in sepsis induce a pro-inflammatory, procoagulant, and proadhesive phenotype in endothelial cells. Under physiological conditions, the endothelium plays a crucial role in vascular homeostasis by expressing antithrombotic and anti-inflammatory mediators such as nitric oxide, prostacyclin (PGI₂), thrombomodulin, and ADAMTS13. Its surface glycocalyx, a negatively charged carbohydrate-rich layer, further contributes to vascular protection by maintaining barrier integrity, responding to shear stress, and preventing leukocyte and platelet adhesion as well as activation of the coagulation cascade [19].
In sepsis, however, the endothelium is directly activated by pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-1), PAMPs, and indirectly by NETs. This leads to degradation of the glycocalyx and increased endothelial permeability due to exposure of the underlying membrane [19].
Protecting the endothelial glycocalyx with aptamers
One of the major challenges in sepsis is to protect and/or restore endothelial function to preserve vascular integrity and limit downstream complications. Glycocalyx degradation is mainly mediated by heparanase, which cleaves heparan sulfate, and ADAM15, which targets CD44. We hypothesize that aptamers directed against these enzymes or their binding sites could preserve glycocalyx integrity. For example, anti-heparanase aptamers developed in oncology have been shown to inhibit tumor cell invasion in in vitro carcinoma models [20]. Similarly, a DNA aptamer targeting the hyaluronic acid-binding domain of CD44 (aptamer s5, K_D_ = 779 nM) inhibited proliferation of leukemia cell lines (in vitro), suggesting possible utility in preventing CD44 cleavage by ADAM15 [21] (Fig. 3).Fig. 3Aptamers targeting endothelial dysfunction in septic DIC. Glycocalyx damage (1) induced vascular permeability and expose endothelium to cytokines, PAMP and immune cells which will activate endothelial cells (2). Afterwards leukocytes and platelets adhere to endothelium (3) and platelets are activated (4). Aptamers targeting these different systems are shown in red. NET: neutrophil extracellular trap, PAMP: pathogen associated molecular pattern, LFA-1: lymphocyte function-associated antigen 1, ICAM-1: intercellular adhesion molecule 1, VCAM-1: vascular cell adhesion molecule 1, vWF: von Willebrand factor, GPVI: glycoprotein VI, PAR: protease-activated receptor.Fig. 3
Targeting endothelial junctions and barrier function
Endothelial glycocalyx disruption leads to exposure of the endothelium and loss of tight and adherens junction integrity. Elevated serum lactate—associated with sepsis severity—disrupts vascular endothelial cadherin, leading to gap formation between endothelial cells and vascular leakage [22]. Inflammatory cytokines or NF-κB pathway activation also reduce the expression of key tight junction proteins (e.g., occludin, ZO-1, claudin-5), facilitating leukocyte transmigration [23]. To date, no aptamers have been developed to directly target endothelial junction proteins such as VE-cadherin, occludin, claudin-5, or ZO-1, but this might represent a potential area for future research aimed at preserving vascular integrity in sepsis.
Aptamers against endothelial activation and leukocyte adhesion
Activated endothelial cells release DAMPs that amplify inflammation and undergo phenotypic changes, including oxidative stress and increased expression of adhesion molecules such as selectins, integrins, ICAM-1, and VCAM-1. These molecules facilitate leukocyte rolling, adhesion, and extravasation. Their circulating levels (ICAM-1, VCAM-1, E-selectin, P-selectin, and vWF) correlate with sepsis severity and poor outcomes [24,25].
Aptamers targeting adhesion molecules represent promising therapeutic tools to block endothelial-leukocyte interactions, limit barrier disruption, and modulate inflammatory signaling. Several aptamers targeting ICAM-1, VCAM-1, LFA-1, as well as P- and E-selectins, have demonstrated functional effects both in vitro and in mouse models of transient cerebral ischemia miming a stoke or sickle cell disease [[26], [27], [28], [29], [30]].
These examples highlight the potential of aptamers to modulate key events in sepsis-induced endothelial dysfunction, thereby reducing vascular leakage, inflammation, and possibly downstream coagulopathy (Fig. 3).
Building on this, another critical therapeutic target in sepsis-induced coagulopathy is the excessive activation of the coagulation cascade, which contributes to microthrombus formation and organ dysfunction.
Targeting dysregulated coagulation pathways
During sepsis, endothelial glycocalyx degradation exposes procoagulant surfaces and increases vascular permeability. Activated or damaged endothelial cells release von Willebrand factor (vWF), promoting platelet adhesion and aggregation. Activated platelets expose phosphatidylserine, which amplifies the intrinsic coagulation pathway. This pathway is also triggered by microvesicles from platelets, immune, and endothelial cells. In parallel, inflammatory stimuli upregulate tissue factor (TF) on immune and endothelial cells, activating the extrinsic pathway and leading to excessive thrombin generation [31]. Elevated thrombin further promotes platelet activation and fibrin formation, contributing to microthrombi.
This hypercoagulable state overwhelms endogenous anticoagulant systems. Antithrombin (AT) and thrombomodulin (TM) levels decrease, resulting in reduced activated protein C (APC), while tissue factor pathway inhibitor (TFPI) becomes insufficient to counteract the procoagulant cascade.
In this context, aptamers could offer a therapeutic strategy by blocking platelet adhesion, inhibiting platelet or coagulation factor activation, or enhancing anticoagulant pathways.
Reducing platelet aggregation and adhesion
During haemostasis, platelets adhere and aggregate at sites of endothelial injury to maintain vascular integrity and prevent bleeding. This process involves adhesion to exposed subendothelial components such as vWF, collagen, fibronectin, and fibrinogen. A potential strategy involves using DNA aptamers targeting in vitro fibronectin to inhibit fibronectin–integrin-mediated adhesion (Fig. 3) [32].
In sepsis and DIC, elevated vWF antigen levels are markers of endothelial activation and disease severity [33]. vWF is normally cleaved by ADAMTS13 into less thrombogenic fragments. However, in sepsis, excessive vWF release consumes ADAMTS13, resulting in a prothrombotic imbalance. Several aptamers targeting vWF have been developed (Fig. 3). ARC1779 binds the A1 domain of activated vWF, blocking its interaction with the GPIb receptor on platelets. It has shown good tolerability with no bleeding complications in a first-in-human trial (NCT00432770) [34]. ARC1779 inhibits all vWF-mediated platelet activation pathways and has been evaluated in five phase II trials (NCT00632242, NCT00726544, NCT00694785, NCT00507338, NCT00742612) for conditions such as thrombotic thrombocytopenic purpura [35,36], von Willebrand disease type 2B [37], acute myocardial infarction [38], and cerebral thromboembolism after carotid endarterectomy [39].
Other vWF-targeting aptamers including BT200, ARC15105, TAGX-0004, R9.3, R9.14, and DTRI-031 also reduce platelet adhesion and clot formation in in vitro, ex vivo and in vivo models (Figure 3) [[40], [41], [42], [43], [44]]. BT200 reduced platelet adhesion and thrombus formation in ex vivo models using healthy volunteers (stimulated with desmopressin or LPS). In parallel, the aptamer DTRI-031 effectively dissolves platelet-rich clots in carotid artery (mouse and canine) and saphenous vein (mouse) thrombosis models, underscoring its potential therapeutic value for thrombosis management in non-pathological settings. These findings highlight the potential of aptamers to disrupt vWF–GPIb-V-IX interactions and limit platelet adhesion.
Moreover, P-selectin aptamers (discussed in Part 2) could inhibit platelet–endothelium adhesion by targeting P-selectin/PSGL-1 interactions (Fig. 3), potentially reducing microthrombus formation in sepsis, although the degree of protection may vary depending on the experimental model and underlying inflammatory context.
In addition to adhesion, platelet activation plays a key role in thrombo-inflammation. Platelets are activated by pathogens via PRRs, GPVI, and FcγRIIA, and by soluble agonists like thrombin (via PAR1/PAR4), ADP (via P2Y1/P2Y12), and thromboxane A₂ (via TP receptor). Activated platelets release granules with prothrombotic and pro-inflammatory contents and shed microvesicles bearing TF and phosphatidylserine.
Aptamers targeting platelet activation pathways may reduce this procoagulant activity. The RB571 aptamer targets GPVI and inhibits collagen-related peptide-induced platelet aggregation, with an IC_50_ of 30 nM in vitro [45] (Fig. 3). Aptamers targeting thrombin exosite I have also been shown to block thrombin–PAR1 interaction and platelet activation [46]. Since activated platelets provide a phosphatidylserine-rich surface for intrinsic coagulation activation, aptamers interfering with platelet activation may offer a targeted anticoagulant effect.
Together, these approaches suggest that aptamers may improve therapeutic outcomes in sepsis-associated coagulopathy by limiting platelet adhesion, activation, and potentially downstream thrombus formation.
Inhibiting factors of the coagulation cascade
A stable blood clot is formed during the second phase of haemostasis through a series of enzyme-driven reactions that stabilize the initial platelet plug. The coagulation cascade is divided into three interconnected pathways: intrinsic, extrinsic, and common.
Intrinsic pathway: FXII and FXI inhibition
Activation of factor XII (FXII) initiates the intrinsic pathway and proceeds via FXI and FIX, ultimately activating FX. Exposed phosphatidylserines on microvesicles interact with FXII, inducing conformational changes that promote its autoactivation (contact activation). This process is enhanced by collagen, kininogen, and proteolytic enzymes such as kallikrein, thrombin, and trypsin. As discussed above, endothelial activation in sepsis promotes a procoagulant and proinflammatory state, contributing to microvascular thrombosis.
Importantly, the FXII/FXI pathway contributes to thrombus growth but is dispensable for physiological haemostasis [47], making it an attractive therapeutic target. The aptamer R4cXII-1 inhibits, in vitro, FXII autoactivation and downstream FXI activation, delaying clot formation. However, it does not block FXIIa activation mediated by the kallikrein–kinin system [48].
In addition, several aptamers have been developed to target in vitro FXIa, including the Factor Eleven Inhibitory Aptamer (FELIAP) [49], and the 11.16 and 12.7 aptamers [50]. These molecules prolong clotting times by approximately 1.3- to 2-fold through inhibition of the intrinsic coagulation pathway, which suppresses thrombin generation and consequently delays clot formation (Fig. 4). Inhibition of FXIa is particularly attractive as it reduces thrombosis while exerting only a limited impact on physiological haemostasis, thereby lowering the risk of bleeding compared to conventional anticoagulants.Fig. 4Aptamers targeting excessive coagulation activation. Excess of coagulation activation (1) and downregulation of endogenous anticoagulants (2) in septic DIC forms stable fibrin clot responsible of organ failure. Inhibitor aptamers are in red whereas agonist aptamer is in green. The coagulation cascade factors are highlighted in cyan. Endogenous anticoagulants are represented in purple. NET: neutrophil extracellular trap, TF: tissue factor, FXIIa: Factor XII activated, AT: antithrombin, TM: thrombomodulin, APC: activated protein C, TFPI: tissue factor pathway inhibitor.Fig. 4
FIXa inhibition: the REG1 system and others
The REG1 system comprises RB006, an RNA aptamer targeting FIXa, and its complementary RNA antidote RB007, allowing precise and reversible anticoagulation. REG1 demonstrated efficacy and safety in phase II clinical trials for acute coronary syndrome (NCT00932100) and coronary artery disease (NCT00715455) [51]. However, phase III development was halted due to severe allergic reactions, likely related to PEG conjugated to the 3' end of RB006 [52].
Despite this, the antidote RB007 showed strong efficacy in restoring clotting times to baseline levels, highlighting the therapeutic potential of aptamer-antidote systems [53]. Additional FIXa-targeting aptamers have been identified. Among these, DTRI-178 was evaluated in a veno-arterial extracorporeal membrane oxygenation (ECMO) model in pigs. This study assessed ECMO-induced coagulopathy and showed that a single dose of aptamer effectively prevented thrombosis. Its anticoagulant efficacy was comparable to that achieved with continuous unfractionated heparin (UFH) infusion. Notably, DTRI-178 was associated with reduced bleeding at the surgical site and a lower transfusion requirement compared with UFH [54]. Overall, these aptamers demonstrated effective anticoagulant activity and expanded the range of FIXa-targeted therapeutic options (Fig. 4).
Extrinsic pathway: TF–FVIIa complex
The extrinsic pathway is activated by TF exposure, typically from damaged tissues. In sepsis, TF is also highly expressed by immune cells, activated endothelium, and platelets, as well as their released microvesicles, contributing to upregulation of the extrinsic coagulation pathway.
The RNA aptamer 16.3 blocks in vitro the formation of the TF–FVIIa complex, thereby preventing FX activation and prolonging prothrombin time by 1.8-fold in human plasma [55]. With a plasma half-life of 15 h in vitro, the aptamer 16.3 is a promising candidate for in vivo use in sepsis-induced coagulopathy, although further studies are needed to establish its pharmacokinetic profile and therapeutic efficacy under physiological and pathological conditions (Fig. 4).
Common pathway: FX, FXa/FVa complex, and thrombin
FX is activated by both the intrinsic (FIXa, with FVIIIa) and extrinsic (TF–FVIIa) pathways. FXa then forms a complex with FVa to convert prothrombin (FII) into thrombin (FIIa), which cleaves fibrinogen into fibrin and activates FXIII to stabilize the clot.
Disrupting the FXa–FVa complex could inhibit thrombin generation independently of upstream pathways. The aptamer T18.3 binds both FV and FVa, displaying anticoagulant activity in plasma from COVID-19 patients [56]. Another strategy is to directly inhibit FX or FXa using RNA11F7t, which prevents activation and interaction with FVIIIa [57]. It was studied ex vivo using healthy human blood circulated through an extracorporeal oxygenator circuit designed to reproduce cardiopulmonary bypass conditions. In this setting, RNA11F7t was used to investigate coagulopathy related to extracorporeal circulation in a surgical, non-infectious context. To further improve efficacy, multimeric aptamers have been developed. The bivalent aptamer RNABA4 combines RNA11F7t and R9D-14T, simultaneously targeting FX/FXa and thrombin. This dual mechanism inhibits in vitro both thrombin generation and fibrin formation, amplifying anticoagulant effects and providing precise control of coagulation [58].
Thrombin inhibition: exosite I, exosite II, and dual targeting
Thrombin, a central enzyme in coagulation, has two exosites: exosite I, which binds fibrinogen, thrombin receptor, and thrombomodulin, and exosite II, which binds heparin and prothrombin domains [59].
The first thrombin-binding aptamer, HD1 (also known as ARC183 or TBA), was discovered in 1992 [60]. It was studied in the cynomolgus monkey in an extracorporeal circulation model [61]. However, its clinical development was discontinued due to high dose requirements [62]. Subsequently, several next-generation aptamers have been designed to target thrombin exosite I with greater affinity and specificity, although most of them have been evaluated only in vitro [[71], [72], [73], [74], [75]]. The aptamer NU172 has been tested in a phase II clinical trial for off-pump coronary artery bypass grafting (NCT00808964) [63]. Another anti-exosite I aptamer, M08 has been prolonging clotting time in healthy mice [63].
Exosite II–binding aptamers, though generally less potent, are valuable for more selective modulation. HD22, one of the earliest, binds exosite II and inhibits activation of coagulation cofactors V and VIII, interfering with clot amplification [64]. HD22 has been studied ex vivo with human blood physiological flow conditions. Other aptamers such as Tog25 also inhibit in vitro clot formation and platelet activation [65] (Fig. 4).
Bivalent aptamers that simultaneously bind exosites I and II show enhanced anticoagulant effects. HD1-HD22 (in vitro) and M08-HD22 (in healthy mice) combine the specificity of HD1 or M08 with HD22 [66,67]. Optimizing linker length and composition produced a synergistic effect, with efficacy up to four times that of individual aptamers [67]. These aptamers disrupt both initiation and amplification phases of thrombin-mediated coagulation, reducing thrombus formation while potentially minimizing bleeding risk.
Counteracting downregulation of endogenous anticoagulants
In sepsis-induced DIC, endogenous anticoagulant systems are significantly downregulated, contributing to the procoagulant state characteristic of DIC. The main anticoagulants affected include antithrombin (AT), thrombomodulin (TM), and tissue factor pathway inhibitor (TFPI).
TM, an endothelial membrane receptor, forms a complex with thrombin to activate protein C (in the presence of protein S), which then inactivates FVa and FVIIIa. In sepsis, endothelial dysfunction leads to the shedding of TM, increasing soluble TM levels in plasma and reducing its availability on the cell surface, resulting in insufficient generation of activated protein C (APC). Moreover, TM downregulation contributes to hyperinflammation, as HMGB1, a key inflammatory mediator usually cleaved and inactivated by the thrombin–TM complex, remains active in septic DIC.
In parallel, TFPI is consumed through its binding to FXa and the TF–FVIIa complex. Inflammatory stimuli also suppress TFPI synthesis in endothelial cells and monocytes. Despite these insights, replacement therapies using exogenous AT, APC, or TM have not improved patient outcomes, and often increased bleeding risks [68].
An alternative strategy would be to counteract the downregulation of these endogenous anticoagulants by enhancing their function or availability, rather than supplementing them directly. This could restore physiological anticoagulant balance without increasing bleeding risk, a key limitation of conventional therapies.
AT is a serine protease inhibitor that neutralizes key coagulation enzymes such as thrombin and FXa. During sepsis, AT levels decrease due to: [1] consumption through formation of irreversible complexes with thrombin and FXa [2], loss of heparan sulfate from the endothelial surface, which is required to activate AT, and [3] degradation by neutrophil elastase during NETosis [69].
Although aptamers are typically used to inhibit targets, they can also act as agonists, binding their targets to induce beneficial conformational changes. AT-16 is an RNA aptamer that specifically binds AT and enhances in vitro its anticoagulant activity by increasing its affinity for FXa (Fig. 4) [70]. By promoting this conformational shift, AT-16 potentiates AT’s inhibition of FXa, reducing thrombin generation and subsequent clot formation.
Unlike heparin, which also activates AT but binds multiple targets (e.g., heparin cofactor II, protein C inhibitor, TFPI), AT-16 exhibits higher specificity and affinity for AT, potentially reducing off-target effects [71]. While the efficacy and safety of the aptamer-mediated enhancement of AT activity in sepsis-induced DIC, particularly regarding bleeding risk, still require confirmation in clinically relevant models, this approach holds promise as a novel strategy to restore anticoagulant balance in this condition.
Restoring defective fibrinolysis in sepsis-induced DIC
Fibrinolysis and coagulation tightly interact to maintain haemostatic balance. The primary role of fibrinolysis is to degrade fibrin networks within clots. Endothelial cells release tissue plasminogen activator (tPA), which converts plasminogen into plasmin, the main enzyme responsible for clot dissolution. In patients with sepsis-induced DIC, fibrinolysis is impaired [72], mainly due to elevated levels of plasminogen activator inhibitor-1 (PAI-1), which blocks both tPA and urokinase-type plasminogen activator (uPA), preventing plasminogen activation and promoting a prothrombotic state [73].
To counteract this, aptamers targeting PAI-1 have been developed. Among them, R10-4 and R10-2 specifically inhibit in vitro the anti-proteolytic activity of PAI-1 against tPA, thereby enhancing fibrinolysis by allowing tPA to more effectively convert plasminogen to plasmin [74] (Fig. 5). These aptamers represent a promising therapeutic strategy in settings where excessive PAI-1 suppresses fibrinolysis. However, their clinical efficacy and safety remain to be demonstrated in vivo, considering the intricate regulation of fibrinolysis and potential off-target effects in septic coagulopathy. In addition to excessive PAI-1, free plasmin that does form in septic DIC is rapidly neutralized by α2-antiplasmin [75]. This leads to the formation of plasmin–antiplasmin complexes, further impairing fibrinolysis. Moreover, FXIIIa, activated by thrombin, stabilizes clots by crosslinking fibrin and incorporates α2-antiplasmin into the fibrin network, protecting it from degradation.Fig. 5Aptamers targeting the fibrinolytic pathway in septic DIC. Aptamers are in red. Fibrinolytic system is shown in coral-colored. Fibrinolysis inhibitors are in grey. Thrombin refers to coagulation pathway occurs in cyan. NET: neutrophil extracellular trap, uPA: urokinase plasminogen activator, tPA: tissue-type plasminogen activator, PAI-1: plasminogen activator inhibitor type-1, α2AP: α2-antiplasmin, α2M: α2-macroglobulin, TAFI: thrombin activatable fibrinolysis inhibitor.Fig. 5
Inhibiting FXIIIa could thus help restore fibrinolysis. In vitro studies showed that aptamers targeting FXIIIa reduce clot formation. Notably, the FA12 aptamer inhibits FXIIIa-mediated incorporation of α2-antiplasmin into fibrin, reducing clot rigidity and increasing susceptibility to lysis [76] (Fig. 5).
Other inhibitors of fibrinolysis include thrombin-activatable fibrinolysis inhibitor (TAFI) and α2-macroglobulin. TAFI, once activated by thrombin, is strongly potentiated (∼1250-fold) by thrombomodulin [77]. Active TAFI cleaves C-terminal lysine residues from fibrin degradation products, removing key binding sites for tPA and plasminogen. This disrupts the positive feedback loop that enhances plasmin generation.
Finally, α2-macroglobulin serves as a secondary inhibitor of plasmin when α2-antiplasmin is depleted. Thus, developing aptamers targeting TAFI, α2-antiplasmin, or α2-macroglobulin could represent promising future strategies to restore fibrinolysis in sepsis-induced coagulopathy.
Targeting neutrophil-derived mediators and NETs to decrease excessive coagulation activation and restore fibrinolysis
Neutrophils play a central role in innate immunity by migrating to sites of infection or injury and exhibiting antimicrobial functions. Their release into peripheral blood is tightly regulated by the CXCR4–CXCL12 axis, but during sepsis, CXCL12 is downregulated, leading to increased neutrophil release [78]. In circulation, neutrophils become more deformable and express integrins that enable margination and adhesion to the endothelium via selectins. Bacterial components in sepsis enhance expression of β1/β2 integrins on neutrophils and upregulate ICAM-1 and VCAM-1 on endothelial cells, promoting firm adhesion. Transmigration to infected tissues is mediated by chemokine–CXCR2 interactions, though CXCR2 is progressively downregulated in later stages of sepsis [79].
Neutrophils release both pro- and anti-inflammatory mediators and are therefore potential therapeutic targets to mitigate excessive coagulation and restore fibrinolysis in sepsis-induced coagulopathy.
One major neutrophil effector mechanism is NETosis, a specialized form of programmed cell death triggered by PAMPs, cytokines, and activated platelets. During NETosis, neutrophil elastase and myeloperoxidase translocate to the nucleus, leading to chromatin decondensation and rupture of the nuclear envelope. The resulting NETs are composed of DNA, histones (H3, H4), and granule proteins (elastase, cathepsin-G, MPO), and function to immobilize and kill pathogens.
While beneficial in infection control, excessive NET formation and impaired clearance contribute to endothelial damage and thrombo-inflammation. During sepsis, NETs adhere to and activate endothelial cells via histone-dependent mechanisms. Histones stimulate TLR pathways, promoting cytokine release and vascular injury.
RNA aptamers targeting histones H3 and H4 have been shown to neutralize their proinflammatory activity in human cells and in a murine model of multiple organ dysfunction induced by intravenous injection of calf thymus histones (Figure 5) [80]. These aptamers may reduce cytokine release, endothelial activation, and NET-associated damage.
NETs also act as a scaffold for platelet adhesion and aggregation, and their DNA/protein content activates FXII, promoting fibrin generation. Moreover, neutrophil elastase, a key NET component, degrades plasminogen, limiting plasmin generation and impairing fibrinolysis [9].
Aptamers targeting neutrophil elastase may help restore fibrinolysis. The RNA aptamer NX21909 inhibits elastase activity in a rat model of acute lung injury induced by tracheal administration of IgG anti-bovine serum albumin (BSA) then BSA intravenous injection, reducing both tissue damage and neutrophil recruitment to the lungs [81] (Fig. 5). By limiting plasminogen degradation and inflammatory infiltration, such aptamers could counteract neutrophil-driven fibrinolytic failure in sepsis.
Another NET-associated protease, cathepsin-G, contributes to extracellular matrix degradation, platelet aggregation, and proinflammatory signaling. While essential in host defense, excess cathepsin-G activity aggravates tissue injury in inflammatory conditions like sepsis. The CG51 aptamer, developed to inhibit cathepsin-G, reduces in vitro its proteolytic activity and may help rebalance immune responses while limiting tissue destruction [82].
These aptamers demonstrate the potential to selectively modulate NET components, reduce thrombo-inflammation, and mitigate endothelial injury. Nevertheless, their efficacy in fully restoring fibrinolysis and preserving endothelial integrity in septic coagulopathy remains to be validated in clinical studies, given the complexity of NET-mediated pathophysiology. Building on these mechanisms, another critical driver of sepsis-induced DIC is the complement system, which tightly interconnects with coagulation and inflammation.
Targeting complement system in sepsis-induced DIC
The complement system and coagulation cascade are closely interconnected. Complement proteins can activate coagulation, while coagulation factors such as thrombin and FXa can directly activate the complement system, establishing a positive feedback loop that contributes to the development of DIC [83]. This process is exacerbated by the downregulation of anticoagulants like thrombomodulin (TM) and activated protein C (APC), which normally inhibit complement activation [84]. Loss of these regulatory mechanisms amplifies both coagulation and inflammation, worsening the clinical course.
Given this interplay, the complement system represents a promising therapeutic target in sepsis-induced DIC. A randomized controlled trial involving C1-inhibitor in 40 patients with severe sepsis or septic shock showed mild benefit in reducing organ dysfunction, likely due to broad inhibition of all three complement pathways [85].
At the center of complement activation is C3, which integrates signals from the classical, lectin, and alternative pathways. The classical pathway is triggered by antibodies and pentraxins (e.g., CRP, serum amyloid P, pentraxin 3), the lectin pathway by mannose-binding lectin recognizing pathogen-associated carbohydrates, and the alternative pathway by spontaneous C3 hydrolysis on foreign or damaged cells.
The alternative pathway is critical for amplifying C3 activation. Factor B is a key mediator that helps form C3 and C5 convertases, essential enzymes in the cascade. Several aptamers have been developed to specifically inhibit in vitro factor B, preventing C3 convertase formation and regulating the alternative pathway (Fig. 6) [86].
Upon activation, complement proteins are cleaved into two fragments: a smaller “a” subunit (anaphylatoxin) and a larger “b” subunit with binding activity. For instance, C3b amplifies its own production, opsonizes pathogens, and participates in C5 cleavage into C5a (a potent proinflammatory mediator) and C5b, which initiates the membrane attack complex (MAC). Excessive MAC formation can lead to tissue injury [87].
C3a and C5a, the main anaphylatoxins, recruit neutrophils and monocytes, increase vascular permeability, and upregulate endothelial adhesion molecules. Elevated levels of these fragments during sepsis have been linked to anaphylactic shock, endothelial damage, and coagulation activation, ultimately contributing to DIC [88,89]. Notably, C5a promotes procoagulant activity by upregulating tissue factor (TF) in endothelial cells and neutrophils, and by shifting mast cells and basophils from a profibrinolytic to a prothrombotic phenotype, via increased PAI-1 expression [[90], [91], [92]].
Aptamers targeting the complement system could offer new therapeutic opportunities (Fig. 6):
- •Avacincaptad pegol (Iverzay™), an FDA-approved RNA aptamer (2023), blocks C5 cleavage into C5a and C5b. This inhibits both C5a-induced inflammation and MAC formation, offering protection from complement-mediated tissue damage. Although approved for geographic atrophy in age-related macular degeneration, its dual anti-inflammatory and cytoprotective action could be valuable in sepsis [93].
- •NOX-D20, a Spiegelmer (L-nucleotide aptamer), binds C5a and competes with its receptor, thereby inhibiting C5a-induced immune responses. Due to its L-nucleotide structure, NOX-D20 is resistant to nuclease degradation, enhancing its stability in vivo. In a murine model of polymicrobial sepsis (cecal ligation and puncture), NOX-D20 reduced inflammation, prevented endothelial barrier breakdown, decreased organ injury, and improved the 7-day survival [94]. Survival did not differ between treatment groups or among the tested dosing regimens (two daily doses of 1 mg/kg and 3 mg/kg, or a single 1 mg/kg dose). Fig. 6Aptamers targeting the complement system exacerbating inflammatory and procoagulant states in septic DIC. Aptamers are in red. Complement system is shown in yellow. Simplified coagulation pathway occurs in cyan. C1: complement 1, MAC: membrane attack complex, TF: tissue factor, FXIIa: Factor XII activated, PAI-1: plasminogen activator inhibitor type-1.Fig. 6
These findings support the therapeutic potential of complement-targeting aptamers in sepsis-induced DIC by simultaneously addressing inflammation, endothelial dysfunction, and coagulation activation. Nevertheless, their clinical efficacy and safety must be evaluated in patients with sepsis-induced coagulopathy to confirm their therapeutic potential.
Aptamers for diagnosis and prognosis tools of sepsis-induced coagulopathy
In addition to their potential therapeutic applications in sepsis-induced DIC, aptamers may also be developed as diagnostic or prognostic tools (Table 3).
First, aptamers can help detect markers of infection. Alongside C-reactive protein, procalcitonin (PCT) is a well-established biomarker for early bacterial infections and is significantly elevated in patients with DIC [95,96]. An aptamer-based biosensor has been developed to detect PCT in clinical samples within 5 min, with a detection limit of 0.01 ng/mL [97]. Compared to conventional methods, this approach is faster, more sensitive, and requires smaller sample volumes.
Aptamers also offer potential in identifying DIC-related markers. Platelet count and fibrinogen are integral to the ISTH DIC score. In septic patients, platelets are often reduced and activated, expressing markers such as P-selectin, CD63, CD31, and soluble GPVI, and contributing to inflammatory aggregates [98]. Aptamers targeting CD63 and CD31 have been developed to isolate specific cell populations or exosomes [99,100]. Similarly, a fibrinogen-specific aptamer has shown high sensitivity in imaging thrombi, even at sub-nanomolar concentrations [101], suggesting utility for real-time thrombotic event monitoring [102].
Endothelial dysfunction can also be explored using aptamer tools. Sphingosine-1-phosphate (S1P), a component of the endothelial glycocalyx that preserves vascular integrity, is inversely associated with sepsis severity [103]. NOX-S93, an aptamer inhibiting S1P's proangiogenic activity, may serve as a prognostic marker [104]. Inflammatory signals in sepsis also induce angiopoietin-2 secretion, promoting glycocalyx degradation via endothelial heparanase [105]. RNA aptamers targeting angiopoietin-2 have been validated in angiogenesis models [106] and may be repurposed for sepsis. Similarly, thrombospondin-1, which modulates coagulation and is released during endothelial activation, has been detected with high sensitivity using aptamer-based colorimetric sensors [[107], [108], [109], [110]]. Finally, hypotension and increased vascular permeability induced by bacteria infection or endotoxemia are due to upregulation of the kinin B1 receptor in vascular tissues and increased bradykinin levels [111,112]. Aptamers targeting the kinin B1 receptor [113], or neutrophil elastase [114,115], may reflect vascular leakage and NETosis-related endothelial injury.
Aptamers could also be used to monitor anticoagulant levels. Protein C is reduced in septic DIC and serves as a prognostic marker [116]. The DNA aptamer HS02-88, initially developed to inhibit APC [117], may be repurposed to quantify APC levels in diagnostic assays. TFPI, another key anticoagulant downregulated in sepsis, is associated with disease severity [118]. While aptamers such as BAX499 and ARC19499 were developed as TFPI inhibitors for bleeding disorders [119,120], they demonstrate the feasibility of aptamer-based modulation and detection of TFPI.
Markers of hypofibrinolysis also deserve attention. tPA and uPA, activators of plasminogen, are inhibited in sepsis mainly by elevated PAI-1. Interestingly, the ProCESS trial found that high circulating tPA levels were associated with increased mortality [121], suggesting that tPA quantification could guide prognosis. The bivalent RNA aptamer 3218 binds tPA with high affinity and could be adapted for measurement [122]. Aptamers targeting uPA—originally developed to block tumor cell invasion [123]—may also have prognostic value, as uPA levels correlate with severity in septic shock [124]. Given PAI-1’s central role in suppressing fibrinolysis [73], aptamers such as R10-2 and R10-4 that block its inhibitory function [74,125] could serve dual roles as therapeutics and diagnostic tools, helping to stratify thrombotic risk in sepsis-induced DIC.
Discussion
Nucleic acid aptamers were recognized as one of the top ten most promising emerging technologies in chemistry in November 2024 [126], and have appeared as a rapidly advancing class of molecules in clinical research. For therapeutic applications [127], 14 aptamers are currently undergoing clinical investigation across 23 Phase I and II trials, for different indications (cancer, cardiovascular disease, viral infections, autoimmune and inflammatory conditions), and two aptamers have received FDA approval: pegaptanib (Macugen®), approved in 2004 to inhibit VEGF in age-related macular degeneration [128]; and avacincaptad pegol (Izervay™), approved in 2023, to target the complement component C5, to treat geographic atrophy in age-related macular degeneration [93].
However, no aptamers have so far reached approval or advanced clinical stages for septic coagulopathy, and only one has been investigated in sepsis-related settings [94]. This limited progress is due to several factors. First, the complex and rapidly evolving pathophysiology of sepsis-induced coagulopathy complicates the development of appropriate preclinical models and hampers clinical translation, particularly because of patient stratification challenges. Other limitations are intrinsic to aptamer therapeutics. The major challenges for aptamer development are associated with their limited in vivo stability, suboptimal delivery efficiency, and intense market competition, particularly from monoclonal antibodies.
In this context, patients with sepsis-induced DIC may typically receive continuous intravenous treatments for supportive care. As a result, administration of an aptamer-based therapy through the same route would be both feasible and compatible with standard intensive care practices. For example, we have recently shown that 50% of anti-thrombin aptamers is conserved during 24 h in septic patient plasmas (unpublished results). Therefore, delivery route and short half-life are not expected to represent a limitation.
Aptamers also benefit from decades of accumulated knowledge in nucleic acid–based therapeutics, recently accelerated by the development of mRNA-based vaccines against SARS-CoV-2 during the COVID-19 pandemic, notably in the area of chemical stabilization. A high number of chemical modifications have been developed and validated for nucleic acids, enhancing in vivo stability, reducing immunogenicity, and improving pharmacokinetics. Advances in nucleic acid therapeutics can be directly applied to aptamer design, increasing the likelihood of developing clinically viable candidates. The diversity of targets and therapeutic potential highlighted in this review indicate that the 55 aptamers described are strong candidates for future clinical application in this field. Consequently, aptamer-based therapies hold considerable promise in the era of precision medicine, offering highly specific and tunable tools to modulate key components of inflammation, endothelial dysfunction, coagulation, and fibrinolysis. In the complex pathophysiology of sepsis-induced coagulopathy, aptamers provide the opportunity to selectively inhibit or enhance molecular targets involved in thrombosis and vascular injury, while minimizing the risk of off-target effects and bleeding complications commonly associated with conventional anticoagulants. Moreover, complementary ‘antidote’ nucleic acid sequences can be used to rapidly reverse aptamer activity (as for example NCT00715455). While the clinical efficacy of these aptamer/antidote systems in septic coagulopathy remains untested, they offer a promising strategy to further reduce side effects such as bleeding complications.
Beyond their therapeutic potential, aptamers show strong promise as diagnostic and prognostic tools through real-time monitoring of biomarkers such as procalcitonin, fibrinogen, neutrophil elastase, or protein C levels. Aptamer-based biosensors [129] in particular, could enable rapid and accurate detection of sepsis and its complications, provided that biomarker relevance in septic coagulopathy is further validated.
Perspectives
Aptamers possess a distinctive set of theoretical and practical advantages, including high target specificity, negligible intrinsic toxicity, reversible activity through complementary antidote sequences, and tuneable pharmacodynamic properties. Despite these advantages, their application in the management of sepsis-induced coagulopathy remains at an early stage, and further comprehensive investigations are required before clinical implementation can be envisaged.
In particular, the establishment of standardized methodologies to assess their therapeutic efficacy will be essential for successful clinical translation. In addition to monitoring the evolution of DIC scores, the efficiency of aptamers cited in this review could be evaluated by measuring soluble markers responsible for endothelial activation, plasma clotting times, markers of fibrinolysis such as plasminogen, D-dimers and fibrin monomers, and plasma activated fragments of complement. Nevertheless, establishing the optimal window for evaluating aptamer efficacy in patients will be a critical step toward clinical validation.
Future research should also aim to confirm the specificity, safety and efficacy of aptamer-based interventions in relevant clinical settings and to evaluate their potential integration into precision-medicine strategies for sepsis-induced DIC. As mechanistic insights into thrombo-inflammatory pathways continue to advance, aptamers may emerge as valuable modulators within a more targeted and individualized therapeutic framework. Notably, their relatively short in vivo half-life, often regarded as a limitation, may confer an advantage in sepsis and septic shock, where rapid and reversible modulation of excessive coagulation may be desirable.
In conclusion, the versatility, high affinity, and chemical adaptability of aptamers make them emerging candidates for both therapeutic intervention and point-of-care diagnostics in critical care. Although they remain a comparatively underexplored class of innovative nucleic acid therapeutics [127], the breadth of potential targets described in this review, and the flexibility of aptamer design provide a promising foundation for innovation in the treatment of sepsis-related coagulopathies.
CRediT authorship contribution statement
Conceptualization and structure, M.M., J.H. and L.C.; formal analysis, M.M.; original draft writing, M.M.; supervision and project administration: J.H. and L.C. All authors have contributed to writing and reviewing. All authors have read and agreed to the submitted version of the manuscript
All the authors have contributed to writing and have revised the manuscript.
Consent for publication
NA.
Ethics approval and consent to participate
NA.
Funding
This research was supported by ADIRAL.
Declaration of competing interest
JH has received honoraria for lectures from Pfizer PFE France, Sanofi Aventis France, Inotrem, MSD, Octapharma and Shionogi and is part of the steering committee from Bayer and AngioDynamics. The other authors have no conflict of interest to declare.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Singer M.Deutschman C.S.Seymour C.W.Shankar-Hari M.Annane D.Bauer M.The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3)JAMA.31582016 Feb 238018102690333810.1001/jama.2016.0287 PMC 4968574 · doi ↗ · pubmed ↗
- 2Rudd K.E.Johnson S.C.Agesa K.M.Shackelford K.A.Tsoi D.Kievlan D.R.Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study The Lancet.395102192020 Jan 1820021110.1016/S 0140-6736(19)32989-7PMC 697022531954465 · doi ↗ · pubmed ↗
- 3Brinkmann V.Reichard U.Goosmann C.Fauler B.Uhlemann Y.Weiss D.S.Neutrophil extracellular traps kill bacteria Science.30356632004 Mar 5153215351500178210.1126/science.1092385 · doi ↗ · pubmed ↗
- 4Delabranche X.Helms J.Meziani F.Immunohaemostasis: a new view on haemostasis during sepsis Ann Intensive Care.712017 Dec 1172919795810.1186/s 13613-017-0339-5PMC 5712298 · doi ↗ · pubmed ↗
- 5Gould T.J.Lysov Z.Swystun L.L.Dwivedi D.J.Zarychanski R.Fox-Robichaud A.E.Extracellular Histones Increase Tissue Factor Activity and Enhance Thrombin Generation by Human Blood Monocytes Shock Augusta Ga.4662016 Dec 6556622740506610.1097/SHK.0000000000000680 · doi ↗ · pubmed ↗
- 6Iba T.Levy J.H.Maier C.L.Helms J.Umemura Y.Moore H.Updated definition and scoring of Disseminated Intravascular Coagulation in 2025: communication from the ISTH SSC Subcommittee on Disseminated Intravascular Coagulation J Thromb Haemost.2025 Apr S 153878362500220 X 10.1016/j.jtha.2025.03.03840216223 · doi ↗ · pubmed ↗
- 7Unar A.Bertolino L.Patauner F.Gallo R.Durante-Mangoni E.Pathophysiology of Disseminated Intravascular Coagulation in Sepsis: A Clinically Focused Overview Cells.12172023 Aug 22212010.3390/cells 12172120 PMC 1048694537681852 · doi ↗ · pubmed ↗
- 8Popescu N.I.Lupu C.Lupu F.Disseminated intravascular coagulation and its immune mechanisms Blood.139132022 Mar 31197319863442828010.1182/blood.2020007208 PMC 8972096 · doi ↗ · pubmed ↗