Comprehensive Clinical, Diagnostic, and In Silico Assessment of a Novel 1p36.33p36.32 Copy Number Variant
Atieh Eslahi, Mir Salar Kahaei, Bita Barazandeh Shirvan, Masoome Alerasool, Sepideh Tabarestani, Razie Rezaie, Narges Hashemi, Javad Akhondian, Mobina Arabi, Farnoosh Ebrahimzadeh, Mehran Beiraghi Toosi, Majid Mojarrad

TL;DR
This paper reports a new 1p36.33p36.32 duplication in a patient with developmental delay and facial dysmorphism, using clinical and bioinformatic methods to understand its impact.
Contribution
The study identifies a novel 1p36.33p36.32 duplication and expands the known phenotypic spectrum of this genomic region.
Findings
A 2.3 MB duplication in the 1p36.33p36.32 region was detected in a patient with developmental delay and facial dysmorphism.
Candidate genes GABRD, DVL1, and GNB1 were implicated in the observed neurodevelopmental and congenital disorders.
Pathway analysis highlighted the '1P36 Copy Number Variation Syndrome' as significantly involved.
Abstract
Clinical manifestations of 1p36.33 duplications vary depending on duplication size. This region is prone to copy number variants associated with diverse phenotypes. We report a novel 1p36.33p36.32 duplication in a patient with developmental delay and facial dysmorphism. The causative duplication was detected by whole‐genome Oligo‐array CGH and confirmed by real‐time PCR. Integrative bioinformatic analyses—including network analysis, phenotype‐driven gene prioritisation, and dosage sensitivity assessment—were performed to explore the molecular basis of the phenotype; we used integrative bioinformatic analyses, including network analysis, phenotype‐driven gene prioritisation, and dosage sensitivity assessment. Assessment of a child with tonic seizures, developmental delay, and dysmorphic facial traits revealed a 2.3 MB gain in the 1p36.33p36.32 region (nucleotide 898,721 to 3,153,945)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
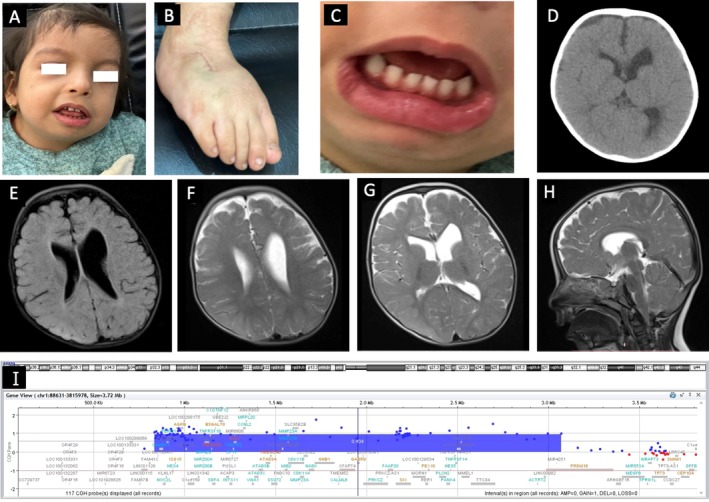 FIGURE 1
FIGURE 1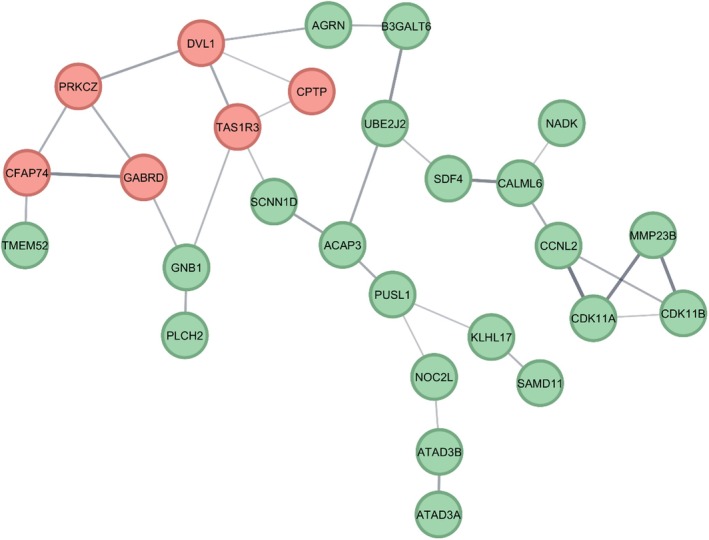 FIGURE 2
FIGURE 2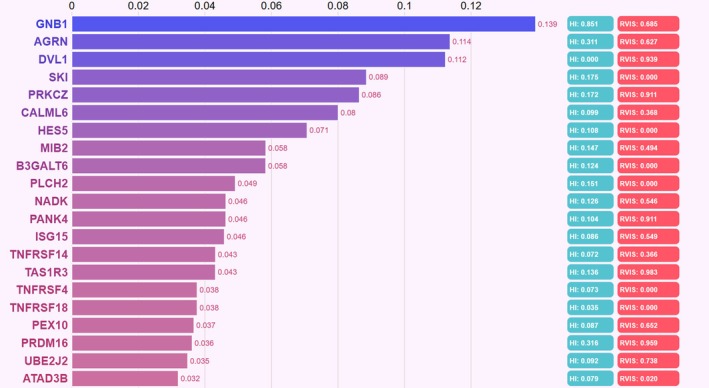 FIGURE 3
FIGURE 3| DECIPHER patient | Sex | Location | Size | Inheritance/genotype | Pathogenicity/contribution | Phenotype(s) |
|---|---|---|---|---|---|---|
| 264,192 | 46XX | 1:875,661–6,076,860 | 5.20 Mb | Unknown | Pathogenic | Generalised‐onset seizure, Global developmental delay, Moderately short stature, Poor speech |
| GRCh38 | Heterozygous | Partial | ||||
| Duplication | ||||||
| 270,940 | 46XY | 1:1,456,616–1,524,663 | 68.05 kb | De novo (parentage confirmed) | Pathogenic | Cardiomegaly, Congenital contracture, Generalised opacification of the cornea, Hypertonia, Hypertrophic cardiomyopathy, Hypospadias, Muscle abnormality related to mitochondrial dysfunction, Overlapping toe, Thoracic scoliosis, Toe clinodactyly |
| GRCh38 | Heterozygous | Full | ||||
| Duplication | ||||||
| 278,905 | 46XY | 1:917,483–5,518,089 | 4.60 Mb | Maternally inherited | Pathogenic | Intellectual disability, moderate |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 331,385 | 46XY | 1:2,112,225–3,227,319 | 1.12 Mb | Maternally inherited | Likely pathogenic | Mild global developmental delay, obsolete Impaired social interactions |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 331,665 | 46XX | 1:2,892,366–3,367,752 | 475.39 kb | De novo (unconfirmed parentage) | Pathogenic | Motor delay |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 331,697 | 46XY | 1:629,046–2,349,810 | 1.72 Mb | Unknown | Likely pathogenic | |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 337,918 | 46XY | 1:824,382–2,563,755 | 1.74 Mb | Unknown | Likely pathogenic | Feeding difficulties, Hypotelorism, Neonatal hypotonia, Retrognathia |
| GRCh38 | Heterozygous | |||||
| Triplication | ||||||
| 394,315 | 46XY | 1:10,001–6,905,674 | 6.90 Mb | Imbalance arising from a balanced parental rearrangement | Likely pathogenic | Abnormal erythrocyte morphology, Broad forehead, Depressed nasal bridge, Hypertelorism, Intellectual disability, Preauricular pit, Ptosis, Small forehead, Strabismus |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 395,321 | 46XX | 1:10,001–6,905,674 | 6.90 Mb | Imbalance arising from a balanced parental rearrangement | Likely pathogenic | Abnormal erythrocyte morphology, Brachydactyly, Hypertelorism, Recurrent infections, Sacral dimple, Strabismus |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 395,323 | 46XY | 1:10,001–6,905,674 | 6.90 Mb | Imbalance arising from a balanced parental rearrangement | Likely pathogenic | Abnormal erythrocyte morphology, Anteverted nares, Broad forehead, Cryptorchidism, Depressed nasal bridge, Hypertelorism, Intellectual disability, Preauricular pit, Ptosis, Small forehead, Strabismus |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 398,345 | 46XX | 1:10,001–6,905,674 | 6.90 Mb | Unknown | Likely pathogenic | 2–3 toe syndactyly, Atrial septal defect, Craniosynostosis, Delayed speech and language development, Intellectual disability, Microcephaly, Narrow forehead, Prominent metopic ridge, Proportionate short stature, Short palpebral fissure, Trigonocephaly |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 400,880 | 46XX | 1:871,284–8,812,156 | 7.94 Mb | Unknown | Likely pathogenic | Abnormal pinna morphology, Brachycephaly, Constipation, Deeply set eye, Delayed closure of the anterior fontanelle, Delayed speech and language development, EEG abnormality, Flat occiput, Frontal bossing, Hypotonia, Intellectual disability, Microcephaly, Midface retrusion, Proportionate short stature, Recurrent infections, Short palpebral fissure, Synophrys, Underdeveloped nasal alae, Wide nasal bridge |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 438,664 | 46XY | 1:3,077,175–5,030,655 | 1.95 Mb | Unknown | Likely pathogenic | |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 472,385 | 46XY | 1:82,154–6,124,132 | 6.04 Mb | Unknown | Pathogenic | |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 472,620 | 46XY | 1:863,579–5,739,571 | 4.88 Mb | Unknown | Pathogenic | |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 486,797 | 46XY | 1:1,354,381–2,542,766 | 1.19 Mb | Paternally inherited | Likely pathogenic | Attention deficit hyperactivity disorder, Delayed puberty, Moderate global developmental delay, Seizure |
| GRCh38 | Heterozygous | Uncertain | ||||
| Duplication | ||||||
| 501,184 | 46XX | 1:787,938–2,400,650 | 1.61 Mb | De novo (parentage confirmed) | Pathogenic | |
| GRCh38 | Heterozygous | |||||
| Amplification | ||||||
| 501,880 | 46XY | 1:2,235,714–3,221,916 | 986.20 kb | Unknown | Likely pathogenic | Specific learning disability |
| GRCh38 | Heterozygous | |||||
| Duplication | ||||||
| 503,091 | 46XX | 1:754,622–3,193,436 | 2.44 Mb | De novo (parentage confirmed) | Likely pathogenic | |
| GRCh38 | Heterozygous | Partial | ||||
| Duplication | ||||||
| 549,911 | 46XY | 1:866,156–3,521,941 | 2.66 Mb | Unknown | Likely pathogenic | |
| GRCh38 | Heterozygous | |||||
| Duplication |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomic variations and chromosomal abnormalities · Genomics and Rare Diseases · Genetics and Neurodevelopmental Disorders
Introduction
1
The 1p36 chromosomal region is susceptible to copy number alterations, encompassing both duplications and deletions, as well as rearrangements. A prevalent genetic anomaly in this area, 1p36 microdeletion, is associated with intellectual disability and developmental delay in infants [1, 2]. Conversely, the less common 1p36 microduplication can result in severe psychomotor delay and speech issues [3, 4]. Copy number variations in this locus correlate with a spectrum of phenotypic features, including hypotonia, spastic paralysis, optic atrophy, cerebellar atrophy, peripheral neuropathy, and syndromic neurological disorders [5]. Additionally, this region harbours genes with diverse copy number variations, such as GABRD, KCNAB2, PLCH2, protein kinase C (PKC)‐zeta, ATAD3, and SKI (v‐ski sarcoma viral oncogene homologue), each contributing to distinct clinical manifestations [6, 7]. For instance, Protein Kinase C (PKC)‐zeta, located in 1p36.33, acts as an atypical member of the PKC family crucial for cell proliferation, survival, and immune response [8]. Therefore, the clinical symptoms associated with the duplication of 1p36.33 seem variable depending on the variation size and the genes involved. Advancements in diagnostic techniques, like array comparative genome hybridisation (array‐CGH), have surpassed traditional karyotyping, enabling the detection of microdeletions, microduplication, and precise breakpoint rearrangements [9]. Here, we highlight a case presenting with neurodevelopmental delay, broad thumbs, esotropia, and a club foot on the right side that had been corrected with surgery. The patient carries a novel heterozygote copy number variant in the 1p36.33 region, as identified by array CGH. Duplication has been confirmed using quantitative Real‐Time PCR confirming a duplication in the PRKCZ gene. To elucidate the potential functional consequences of this duplication, we performed a comprehensive bioinformatic analysis, including protein–protein interaction network construction (STRING, MCODE), phenotype‐driven gene prioritisation (PhenoLyzer), assessment of gene dosage sensitivity (DECIPHER), and pathway enrichment analysis (Enrichr). Additionally, to identify OMIM‐morbid genes within the duplicated region, we used genescout. This integrative approach aimed to identify the candidate genes and biological pathways contributing to the observed phenotype.
Methods
2
Subject and Blood Sampling
2.1
We present clinical features and molecular findings in a 2‐year, 10‐month‐old female with copy number variation in 1p36 that were not previously described in the literature. The patient's peripheral blood was collected into an EDTA tube for genomic DNA extraction using Simbiolab kit. The quantity of DNA in the sample was determined using Nanodrop spectrophotometer. Additionally, agarose gel electrophoresis was used for the assessment of DNA quality. Informed consent has been taken from the patient's parents, allowing to publishing of the patient's clinical and genetic data.
Copy Number Variation Analysis Using Array CGH
2.2
The SurePrint G3 ISCA V2 8X60K whole‐genome Oligo‐Array version 2 (Agilent Technologies, Santa Clara, CA) was employed for whole‐genome oligonucleotide‐based array comparative genomic hybridisation (oligo‐array CGH) analysis. This array comprises approximately 60,000 oligonucleotide probes with an overall median probe spacing of 60 kb across the genome to 500 targeted disease regions. Genomic DNA from the patient and a male reference sample were labelled with Cy5 and Cy3 dyes, respectively, using the Agilent SureTag DNA Labeling Kit (Agilent Technologies) according to the manufacturer's instructions. Following the manufacturer's protocol, the patient's DNA was hybridised against the male reference sample (9399). After hybridisation, the arrays were washed using Agilent wash buffers and scanned using the Agilent SureScan Microarray Scanner (Agilent Technologies) at a resolution of 3 μm. The resulting images were processed using Agilent Feature Extraction software (version 12.0.3.1) to obtain the raw data files.
The raw data files were analysed using Agilent CytoGenomics software (version 4.0.3.12) with the following settings: ADM‐2 algorithm, threshold of 6.0, and a minimum of 4 consecutive probes for called intervals. The log2 ratio of patient/reference signal intensities was calculated, and copy number variations (CNVs) were identified based on the log2 ratio values and their statistical significance.
Quantitative Real‐Time PCR Validation
2.3
To validate copy number changes in the 1p36 region, relative quantitative real‐time PCR (qPCR) assays were conducted using the PRKCZ (protein kinase C, zeta) gene as the target and the KMT2D as the reference gene. The primers were designed with Primer3 software (version 0.4.0) and their sequences were aligned against the entire human genome using the UCSC program (Table S1). The qPCR reactions were set up in 20 μL volumes containing SYBR Green PCR Master Mix (2×), 0.25 μM of each primer, nuclease‐free water, and DNA template (50 ng/reaction). The thermal cycling conditions included an initial denaturation at 95°C for 15 min, and then 35 cycles of 95°C for 30 s and 60°C for 30 s. The qPCR assays were performed using MicPCR equipment. In addition to the test samples, a negative control sample from a normal individual without abnormality in 1p36 was included.
In Silico Analysis
2.4
To systematically prioritise candidate genes within the duplication region identified by array‐CGH, we performed a multi‐step bioinformatics analysis:
Protein–Protein Interaction (PPI) Network Analysis
2.4.1
To comprehensively characterise the duplicated region, we first identified all genes located within the interval using the Ensembl genome browser [10], focusing specifically on protein‐coding genes. Protein interaction networks for these genes were generated using the STRING database [11] and visualised in Cytoscape (v3.10.3) [12], with MCODE [13] applied to detect hub genes and densely connected modules, highlighting potential driver genes within the region.
OMIM Morbid Gene Identification
2.4.2
Genescout [14] was used to systematically identify OMIM‐morbid genes within the duplicated region.
Phenotype‐Driven Gene Prioritisation
2.4.3
To assess genotype–phenotype correlations, we utilised Phenolyzer [15] by providing patient‐specific phenotypes (HPO terms), allowing prioritisation of genes most likely associated with the clinical features observed (Table S2).
Assessment of Gene Dosage Sensitivity and CNV Phenotype Annotation
2.4.4
The DECIPHER database [16] was queried to identify genes within the duplicated region that have documented triplo‐sensitivity or dosage sensitivity. In addition, all reported clinically relevant CNVs overlapping the duplicated region were filtered and systematically reviewed. Associated phenotypic features for each CNV were extracted and summarised in a comparative table.
Pathway Enrichment Analysis
2.4.5
Enrichr [17] was used for pathway enrichment analysis of protein‐coding genes in the duplicated region, focusing on WikiPathways 2024 Human and other relevant pathways.
Results
3
Clinical Evaluation
3.1
The patient, a 2‐year, 10‐month‐old female, was born in a non‐consanguineous family. The parents had two other healthy children, with no history of abortion. The patient exhibited distinctive features including micrognathia, pronounced lower lip, bulbous nose, ample cheeks, widely spaced teeth, a unilateral club foot affecting the right side, and esotropia (Figure 1A–C). At 6 months, she experienced her first tonic seizure that recurred at 2 years, 9 months. The patient's birth weight was 2700 g, last investigated weight was 10 kg, and head circumference was 44 cm. At the age of 2 years, 10 months, she could only crawl with poor social skills. Her phonetic and phonological abilities were limited to crying and babbling, although her social attention was relatively preserved. The cause of the neurodevelopmental delay and tonic seizures was determined by brain MRI and CT, EEG, echocardiogram, and laboratory testing. Laboratory tests revealed normal organic acid levels in urine, while a brain CT scan and MRI showed left ventriculomegaly with a mild abnormality of cortical gyration in the frontal region (Figure 1D–H). Echocardiography showed no abnormalities, and EEG indicated scattered nonspecific sharp waves.
Clinical photographs, brain MRI and CT scan, and array‐CGH results of the patient with 1p36.33 duplication. Clinical photographs showing facial dysmorphism (A), surgically corrected right clubfoot (B), and widely spaced teeth (C). The axial brain CT scan shows left‐side ventriculomegaly without any calcification (D), and the T1 (E) and T2 (F, G, H) sequences of brain MRI show ventriculomegaly especially on the left side with a mild abnormality of cortical gyration on the frontal region. Array‐CGH results indicate a duplication at the 1p36.33p36.32 (GRCh37/hg19; chr1:834,101–3,070,509) (I); genomic coordinates elsewhere in the manuscript are reported on (GRCh38/hg38).
CNV Analysis Using Array CGH and PCR Validation
3.2
Molecular cytogenetic investigation using Array‐CGH revealed a 2.3 Mb duplication CNV at the region 1:898,721–3,153,945 (2.3 Mb), localised in (1p36.33‐p36.32) (Figure 1I). According to ACMG/ClinGen CNV interpretation guidelines [18], the variant met criteria 1A, 2B, 3C, and 4L, leading to its classification as pathogenic. The duplication encompassed 63 protein‐coding genes and partially overlapped an established ClinGen triplosensitive genomic region (ISCA‐37434, 1p36 terminal region). Consistent with the CNV gain scoring metric, this overlap was captured as Evidence 2B (Table S3). Case–control data demonstrated enrichment among affected individuals with consistent phenotypes, and no overlap was detected with common population variants in gnomAD or DGV. Detailed ACMG/ClinGen scoring is provided in Table S3. Real‐time PCR results on the PRKCZ gene confirmed Array‐CGH results.
In Silico Approaches to Array‐Based Candidate Genes
3.3
Network‐Based Identification of Key Genes in a Disease Relevant CNV Region
3.3.1
A total of 89 genes were identified within the duplicated region using Ensembl (hg38), of which 63 were protein‐coding. These 63 protein‐coding genes were subjected to protein–protein interaction (PPI) network analysis using the STRING database. The initial PPI network included several genes with no significant interactions; these disconnected nodes were excluded from further analysis. The final PPI network comprised 27 interconnected nodes and 32 edges, with an average node degree of 2.37, a network diameter of 11, and a clustering coefficient of 0.185. Following PPI network construction, the MCODE algorithm was applied in Cytoscape to identify network modules and hub genes within the connected component (MCODE score = 2.800). MCODE analysis highlighted a cluster of highly interconnected genes (hub genes), including DVL1, PRKCZ, TAS1R3, GABRD, CFAP74, and CPTP (Figure 2). These hub genes may play central roles in the functional network of the duplicated region. According to the Scout gene database, the duplicated region contains 19 OMIM morbid genes, including 9 with autosomal recessive (AR) and 10 with autosomal dominant (AD) inheritance patterns. Among the hub genes identified in the PPI network (CFAP74, GABRD, PRKCZ, DVL1, TAS1R3, CPTP), only CFAP74, GABRD, and DVL1 are classified as OMIM morbid genes. Of these, GABRD and DVL1 are exclusively associated with autosomal dominant inheritance patterns, further supporting their relevance as candidate genes in the context of heterozygous duplications.
Protein–protein interaction (PPI) network of the 27 protein‐coding genes within the duplicated region, constructed using STRING and visualised in Cytoscape. Nodes represent genes; edges represent predicted interactions. Hub genes identified by MCODE are highlighted in red.
Exploring CNV Using Decipher
3.3.2
We systematically reviewed all reported duplication CNVs overlapping this region. Based on DECIPHER data (hg38), a total of 373 copy number and sequence variations—including 59 SNV/indels (less than 50 bp), 218 deletions, and 89 duplications—have been reported within this region. Among the duplications, 20 CNVs are classified as pathogenic or likely pathogenic, and 34 as variants of uncertain significance (VUS). The smallest pathogenic or likely pathogenic CNV reported in this region is 68.05 kb, while the largest reaches 7.94 Mb. Notably, three pathogenic CNVs are less than 1 Mb in size and include the morbid genes PANK4, PEX10, PRDM16, SKI, ATAD3A, CFAP74, GABRD, GNB1, TMEM240, and VWA1. The smallest pathogenic duplication encompasses three genes—ATAD3A, ATAD3B, and ATAD3C—with ATAD3A recognised as a morbid gene in OMIM. Table 1 provides a comparative summary of phenotypes associated with each duplication CNV.
Prioritisation of Candidate Genes and Triplosensitivity Analysis
3.3.3
Using Phenolyzer, we analysed 13 patient phenotypes together with the 89 genes located in the duplicated region to identify the genes most strongly associated with the observed clinical features. This analysis prioritised GNB1 as the top candidate gene (Figure 3). Furthermore, triplosensitivity assessment in the DECIPHER database revealed that among all genes within the duplicated interval, GNB1 was the only gene with a high and significant triplosensitivity score (pTriplo = 1.00). None of the other genes in this region showed a comparable level of predicted triplosensitivity, highlighting GNB1 as the principal dosage‐sensitive gene and supporting its potential relevance to the observed phenotype in our patient.
Phenolyzer‐based prioritisation of genes within the duplicated region using 13 patient‐specific phenotypes (HPO terms). Bars represent the Phenolyzer prioritisation scores for each gene. GNB1 received the highest score among all 89 candidate genes, suggesting its strong association with the observed clinical features. HI, Haploinsufficiency score; RVIS, Residual Variation Intolerance Score.
Pathway Enrichment Analysis
3.3.4
To further investigate the biological relevance of the duplicated region, we performed pathway enrichment analysis using Enrichr. The list of 89 genes identified within the 1p36 duplicated region (based on Ensembl, hg38) was submitted to the Enrichr platform. Using the WikiPathways 2024 Human database, significant enrichment was observed for the ‘1P36 Copy Number Variation Syndrome’ pathway (WP5345), with an adjusted p‐value of 6.420 × 10 ^−111^. Interestingly, the ‘1P36 Copy Number Variation Syndrome’ pathway (WP5345), which is primarily defined based on studies of 1p36 deletions, was also significantly enriched in our gene list. This indicates that the genes duplicated in our case overlap with those implicated in the well‐characterised deletion syndrome; however, the biological and clinical consequences of duplication may differ from those of deletion, highlighting the need for further investigation into dosage effects within this region.
Use of AI‐Assisted Tools in Writing
3.4
We used ChatGPT for language editing and clarity improvements in early drafts of the manuscript. All AI‐generated suggestions were reviewed, verified, and edited by the authors, who take full responsibility for the content. No AI tools were used for study design, data collection, statistical analysis, figure/table generation, or drawing scientific conclusions, and no patient‐identifiable or confidential data were entered into the tool.
Discussion
4
In this report, according to the best of our knowledge, we present the first Iranian case of pathogenic gain of 2.3 Mb resulting in developmental delay and dysmorphic features. This anomaly was identified using array CGH and validated through quantitative Real‐Time PCR, marking the first such case as reported in the DECIPHER database. The child displayed distinctive physical characteristics, encompassing micrognathia, pronounced lower lip, bulbous nose, ample cheeks, widely spaced teeth, unilateral club foot, and esotropia. Additionally, left ventriculomegaly, as observed in the MRI images, was noted as a non‐specific brain feature. Genotype–phenotype correlation in 1p36 CNVs is further complicated by phenotypic heterogeneity. In addition to non‐specific findings such as developmental delay and microcephaly, duplications in this region have been associated with unique features including craniosynostosis, epilepsy, and various dysmorphisms [19, 20, 21, 22]. Moreover, similar clinical manifestations have been reported in cases with deletions, duplications, and even tetrasomy of 1p36 in the DECIPHER database, reflecting the locus's high functional complexity. Therefore, we identified several candidate genes including GABRD, DVL1, and GNB1 through integrative bioinformatic analyses that are likely to contribute to the observed clinical phenotype. GABRD encodes the delta (δ) subunit of the gamma‐aminobutyric acid (GABA) type A receptor, a major inhibitory neurotransmitter receptor in the central nervous system (CNS) (NCBI Gene ID: 2563) [23]. Heterozygous mutations in GABRD have been linked to a range of neurological and psychiatric disorders, most notably Generalised Epilepsy with Febrile Seizures Plus (GEFS+, OMIM #613060). Recent studies have challenged the traditional view that increased GABAergic inhibition is universally protective against seizures. δ‐Containing GABA‐A receptors, encoded by GABRD, mediate tonic inhibition, and gain‐of‐function variants in GABRD have been shown to paradoxically promote absence seizures, both in human patients and animal models [24]. These variants lead to increased tonic GABAergic currents in thalamocortical neurons, altering network excitability and facilitating seizure generation. Similar clinical and electrographic features are observed in patients with loss‐of‐function SLC6A1 variants, which also elevate ambient GABA and enhance δ‐GABA‐A receptor activation [24]. Given that gain‐of‐function and increased dosage of GABRD can lead to enhanced tonic inhibition and seizure susceptibility, it is plausible that the duplication of this gene in our patient may underlie some of the observed clinical features, including epilepsy.
DVL1 belongs to a family of highly conserved proteins (DVL1, DVL2, DVL3) that are key mediators of the Wnt signalling pathway. These proteins act as intracellular scaffolds immediately downstream of Wnt receptors and determine the directionality of Wnt signalling into canonical and non‐canonical pathways. While functional redundancy exists among the paralogs, animal models show that Dvl2 and Dvl3 are critical for development, with loss leading to major congenital anomalies, whereas Dvl1 loss has milder effects [25]. Heterozygous truncating mutations in DVL1 cause autosomal dominant Robinow syndrome‐2 that is a skeletal dysplasia characterised by distinctive facial features, including midface hypoplasia, hypertelorism, a short nose, and a broad mouth, known collectively as fetal facies. Additional features include mesomelic dwarfism, macrocephaly, gingival hypertrophy, dental malocclusion, genital hypoplasia, and brachydactyly [25, 26, 27, 28, 29]. In vitro cellular expression studies demonstrated that co‐transfection of the mutant and wildtype DVL1 resulted in increased activation of the canonical WNT signalling pathway, suggesting a gain‐of‐function effect [25]. These findings indicate that pathogenic DVL1 variants can aberrantly activate WNT signalling even in the presence of a normal allele. Therefore, it is plausible that DVL1 duplication may similarly lead to increased gene dosage and excessive WNT pathway activation, thereby contributing to the clinical phenotype observed in our patient. This underscores the critical importance of DVL1 dosage and WNT pathway regulation in neurodevelopmental and dysmorphic features. GNB1 encodes the beta subunit of heterotrimeric guanine nucleotide‐binding (G) proteins, which act as binary molecular switches in signal transduction [30]. Pathogenic variants in GNB1 cause Intellectual Developmental Disorder, Autosomal Dominant‐42 (OMIM #616973), characterised by global developmental delay and intellectual impairment. Additional features may include hypotonia, seizures, strabismus, cortical visual impairment, and autistic traits. While previous studies have established that loss‐of‐function mutations in GNB1 cause global developmental delay, epilepsy, and a range of neurodevelopmental phenotypes by disrupting G protein signalling [31], the clinical consequences of GNB1 duplication or increased gene dosage remain largely unexplored. Therefore, our findings extend the current knowledge by suggesting that increased GNB1 dosage may also contribute to neurodevelopmental disorders, possibly through a distinct mechanism involving enhanced G protein signalling or dosage imbalance. The observed phenotype in 1p36 duplications may result from the effect of a single gene or from the combined interaction of multiple genes within the duplicated region. For example, GABRD is absent in the smallest duplications and these patients do not exhibit seizures; however, seizures are observed in cases where larger duplications include GABRD, indicating a gene‐specific contribution. In addition to size‐ and content‐dependent effects, the genomic architecture and mechanistic consequences of a copy number variation (CNV) can be critical determinants of pathogenicity. Notably, chromosome 1p36.33 duplication syndrome (OMIM: 618815), involving the ATAD3 gene cluster, overlaps with the CNV interval identified in our patient. This locus lies within a segment of high sequence homology that is prone to non‐allelic homologous recombination (NAHR). That study described highly homologous interval within the ATAD3 region that can serve as the recombination junction, generating an ATAD3A–C fusion gene. Clinically, reported cases exhibit a severe neonatal multisystem phenotype characterised by cardiomyopathy, corneal opacities/clouding, hypotonia, seizures, white‐matter abnormalities (often accompanied by metabolic disturbances such as lactic acidosis), and early lethality [19]. In contrast, our proband's clinical presentation showed limited phenotypic concordance with the classic ATAD3‐cluster duplication phenotype described in the literature. Mechanistically, the same study proposes that the fulminant neonatal phenotype is driven predominantly by a dominant‐negative effect of the ATAD3A–C fusion protein generated by these recurrent NAHR‐mediated duplications, emphasising that duplication architecture (i.e., whether a pathogenic fusion is created) is critical for pathogenicity, with increased ATAD3B dosage alone unlikely to explain the severe neonatal disease.
Taken together, these observations suggest that CNV‐associated phenotypes are not determined by size alone. In some cases, a single key gene may act as the primary driver, with clinical features largely reflecting disruption or altered dosage of that gene. However, even relatively small CNVs can differ markedly in clinical impact depending on the mechanism of formation and the resulting pathogenic architecture—for example, whether the rearrangement is fusion‐forming and capable of a dominant‐negative effect, versus producing a more conventional dosage change. As CNV size increases, additional genes may be included, and phenotypic variability may emerge from gene–gene interactions and pathway‐level perturbations, where the combined effects of multiple dosage changes modulate the ultimate clinical outcome.
Conclusion
5
Our findings highlight the complexity of genotype–phenotype correlations in 1p36 duplications and underscore the importance of combining genomic data with clinical and computational tools for accurate interpretation of rare copy number variants. Further studies, including functional validation and larger cohorts, are warranted to better elucidate the pathogenic mechanisms and improve clinical management of patients with 1p36 duplications.
Author Contributions
Conceptualization: Bita Barazandeh Shirvan, Javad Akhondian, Mehran Beiraghi Toosi, Majid Mojarrad. Methodology: Bita Barazandeh Shirvan. Software: Mir Salar Kahaei. Investigation: Atieh Eslahi, Masoome Alerasool, Javad Akhondian, Mobina Arabi, Farnoosh Ebrahimzadeh, Mehran Beiraghi Toosi. Validation: Narges Hashemi. Formal analysis: Mir Salar Kahaei. Visualisation: Mir Salar Kahaei. Resources: Atieh Eslahi, Majid Mojarrad. Supervision: Mehran Beiraghi Toosi, Majid Mojarrad. Writing – original draft: Bita Barazandeh Shirvan, Sepideh Tabarestani. Writing – review and editing: Razie Rezaie.
Funding
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1: The sequence of primers used for Quantitative Real‐Time PCR. Table S2: List of 13 clinical phenotypes (HPO terms) used as input for gene prioritisation in Phenolyzer analysis. The corresponding HPO identifiers are provided for each phenotype. Table S3: ACMG/ClinGen scoring.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. C. Carey , A. Battaglia , D. Viskochil , and S. B. Cassidy , eds., Cassidy and Allanson's Management of Genetic Syndromes, 1st ed. (Wiley, 2021), cited August 4, 2025, https://onlinelibrary.wiley.com/doi/book/10.1002/9781119432692.
- 2M. Gajecka , K. L. Mackay , and L. G. Shaffer , “Monosomy 1p 36 Deletion Syndrome,” American Journal of Medical Genetics. Part C, Seminars in Medical Genetics 145C, no. 4 (2007): 346–356, https://onlinelibrary.wiley.com/doi/10.1002/ajmg.c.30154.17918734 10.1002/ajmg.c.30154 · doi ↗ · pubmed ↗
- 3A. Battaglia , H. E. Hoyme , B. Dallapiccola , et al., “Further Delineation of Deletion 1p 36 Syndrome in 60 Patients: A Recognizable Phenotype and Common Cause of Developmental Delay and Mental Retardation,” Pediatrics 121, no. 2 (2008): 404–410, https://publications.aap.org/pediatrics/article/121/2/404/68689/Further‐Delineation‐of‐Deletion‐1p 36‐Syndrome‐in.18245432 10.1542/peds.2007-0929 · doi ↗ · pubmed ↗
- 4E. Chen , E. Obolensky , K. A. Rauen , L. G. Shaffer , and X. Li , “Cytogenetic and Array CGH Characterization of De Novo 1p 36 Duplications and Deletion in a Patient With Congenital Cataracts, Hearing Loss, Choanal Atresia, and Mental Retardation,” American Journal of Medical Genetics. Part A 146A, no. 21 (2008): 2785–2790, https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.32437.18924166 10.1002/ajmg.a.32437 · doi ↗ · pubmed ↗
- 5T. Harel , W. H. Yoon , C. Garone , et al., “Recurrent De Novo and Biallelic Variation of ATAD 3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes,” American Journal of Human Genetics 99, no. 4 (2016): 831–845, https://linkinghub.elsevier.com/retrieve/pii/S 0002929716303305.27640307 10.1016/j.ajhg.2016.08.007PMC 5065660 · doi ↗ · pubmed ↗
- 6J. A. Rosenfeld , J. A. Crolla , S. Tomkins , et al., “Refinement of Causative Genes in Monosomy 1p 36 Through Clinical and Molecular Cytogenetic Characterization of Small Interstitial Deletions,” American Journal of Medical Genetics. Part A 152A, no. 8 (2010): 1951–1959, https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.33516.20635359 10.1002/ajmg.a.33516 · doi ↗ · pubmed ↗
- 7N. Bahi‐Buisson , E. Guttierrez‐Delicado , C. Soufflet , et al., “Spectrum of Epilepsy in Terminal 1p 36 Deletion Syndrome,” Epilepsia 49, no. 3 (2008): 509–515, https://onlinelibrary.wiley.com/doi/10.1111/j.1528‐1167.2007.01424.x.18031548 10.1111/j.1528-1167.2007.01424.x · doi ↗ · pubmed ↗
- 8P. Martin , “Role of zeta PKC in B‐Cell Signaling and Function,” EMBO Journal 21, no. 15 (2002): 4049–4057, http://emboj.embopress.org/cgi/doi/10.1093/emboj/cdf 407.12145205 10.1093/emboj/cdf 407PMC 126153 · doi ↗ · pubmed ↗