Gap-less and haplotype-resolved genomes of two Hippophae rhamnoides subspecies: Hippophae rhamnoides subsp. mongolica and Hippophae rhamnoides subsp. sinensis
Zhi-Wei Wang, Ye Zhao, Peng Li, Longxin Wang, Kai-Hua Jia, Cheng-Jiang Ruan

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
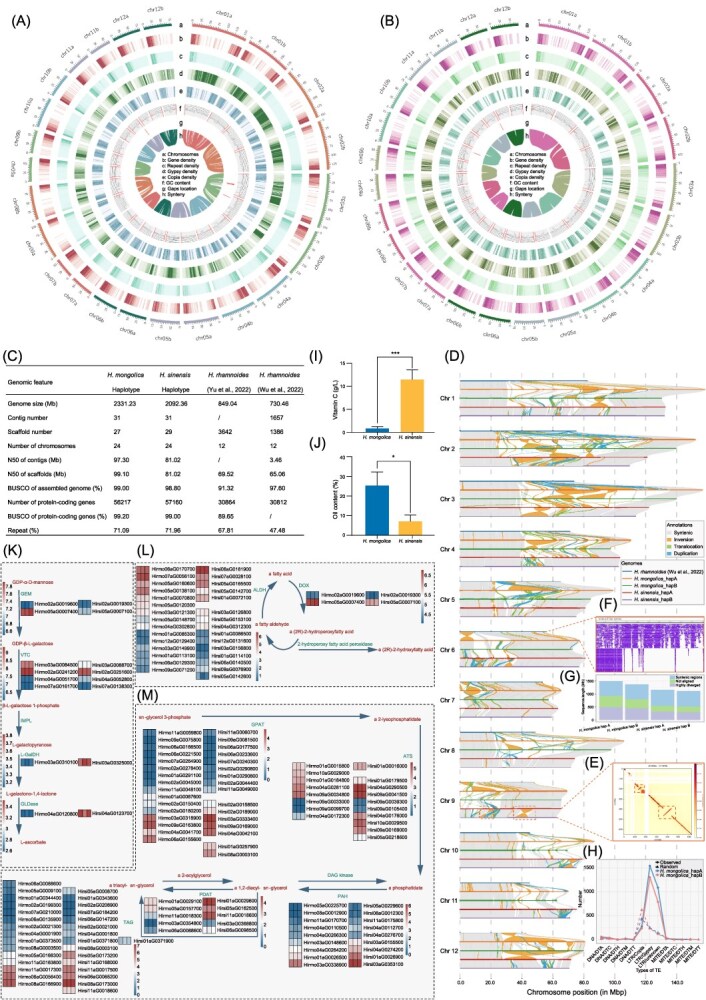 Figure 1
Figure 1- —Fundamental Research Funds for the Central Universities10.13039/501100012226
- —Project of the Sea Buckthorn Development and Management Center of the Ministry of Water Resources
- —International Cooperation Plan Project of Liaoning Province
- —International (Regional) Cooperation and Exchange
- —National Natural Science Foundation of China10.13039/501100001809
- —International (Regional) Collaborative Research
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhytochemical and Pharmacological Studies · Transportation Systems and Logistics · Genetic diversity and population structure
Dear Editor,
Seabuckthorn (Hippophae rhamnoides; 2n = 2x = 24), a member of the Elaeagnaceae family, is native to Asia and Northwestern Europe. This species is renowned for its exceptional resilience to extreme environmental conditions, including temperatures ranging from −40°C to +40°C, drought, and waterlogging, making it highly adaptable for cultivation. Its berries are rich in nutrients and bioactive compounds, notably high levels of flavonoids and vitamin C (Vc), which contribute to its widespread domestication and global cultivation. Recent advancements in sequencing technologies and genome assembly methods have enabled the successful decoding of numerous chromosome-level genomes, some even achieving gap-less or gap-free resolution [1]. Notably, several seabuckthorn genomes have been assembled at the chromosome level, including Hippophae rhamnoides subsp. mongolia (H. r. subsp. mongolica) [2], Hippophae tibetana [3], H. rhamnoides subsp*. sinensis (H. r.* subsp. sinensis) [4], and Hippophae gyantsensis[5]. However, the presence of numerous gaps and unplaced contig sequences poses significant challenges, potentially impacting downstream data analysis and functional research.
In this study, we assembled gap-less, haplotype-resolved genomes of two representative seabuckthorn subspecies H. r. subsp. mongolica which is characterized by large fruits, high yield, high oil content, and sparse thorns; and H. r. subsp. sinensis, which exhibits strong stress resistance, rapid growth, and high Vc content. For H. r. subsp. mongolica, we sequenced 37 Gb of PacBio HiFi reads (30× coverage), 67 Gb of Oxford Nanopore Technologies (ONT) reads (54× coverage), and 73 Gb of Hi-C reads. For H. r. subsp. sinensis, a similar strategy was employed, yielding 37 Gb of PacBio HiFi reads (35× coverage), 61 Gb of ONT reads (60× coverage), and 83 Gb of Hi-C reads. We utilized hifiasm (v0.19.8-r602) to assemble PacBio HiFi and ONT reads into contigs. Hi-C reads were subsequently aligned to the haplotype-resolved assemblies using Juicer (v1.6), and chromosome scaffolding using 3D-DNA (v180419). Manual inspection and correction of chromosome boundaries and misassemblies were performed using Juicebox (v1.11.08). Gaps were filled using quarTeT (v1.2.5) based on HiFi reads. For HiFi reads aligning to chromosome ends, we reassembled these reads into contigs to extend the chromosome lengths as much as possible, aiming to capture the complete telomere sequences. Additionally, GetOrganelle (v1.7.7.1) was employed to assemble the chloroplast and mitochondrial genomes.
After removing redundant sequences, 12 pairs of chromosomes along with chloroplast and mitochondrial genomes were successfully assembled. The genomes of H. r. subsp. mongolica and H. r. subsp. sinensis contained 4 and 2 gaps, respectively, with 38 and 42 telomeres assembled (Fig. 1A-B). The haplotype-resolved assemblies for H. r. subsp. mongolica and H. r. subsp. sinensis were 2.33 Gb and 2.09 Gb, respectively, with contig N50 values of 97 Mb and 81 Mb. For H. r. subsp. mongolica, 99.55% of the PacBio HiFi reads mapped back to the assembly, and 99.76% of the genome was covered by at least 5× HiFi depth. The final genome assemblies exhibited high completeness, with BUSCO (v5.8.2) analysis in genome mode yielding 99.0% complete BUSCOs for H. r. subsp. mongolica and 98.8% for H. r. subsp. sinensis. Hi-C contact maps for both assemblies exhibited clear diagonal blocks without obvious mis-joins, indicating high chromosomal accuracy. These results demonstrate that we have achieved two gap-less haplotype-resolved genomes assemblies. In addition, we assessed phasing quality using multiple complementary approaches, including K-mer spectrum analysis and quantitative evaluation of switch errors. K-mer analysis verified the accuracy of the assembly results, indicating that the sequence differences between haplotypes were clearly distinguished without significant contamination or mosaicism. Switch-error rates were 0.8% for H. r. subsp. mongolica and 0.6% for H. r. subsp. sinensis, both well below the 1% threshold commonly accepted for high-quality phased assemblies. Compared to previously reported seabuckthorn genomes, our assemblies exhibit markedly improved contiguity, with contig N50 values increased by over 20-fold and a substantial reduction in scaffold numbers (Fig. 1C).
Using EDTA (v1.9.9) followed by RepeatMasker (v4.1.8), we annotated 71.09% of the H. r. subsp. mongolica genome and 71.96% of the H. r. subsp. sinensis genome as repetitive. In both assemblies, long terminal repeats (LTRs) dominate: they account for 44.74% of H. r. subsp. mongolica and 46.58% of H. r. subsp. sinensis. Within the LTR fraction, LTR/Gypsy elements are the largest component—23.44% in H. r. subsp. mongolica and 12.56% in H. r. subsp. sinensis. The marked difference in LTR/Gypsy representation is explained by the proportion of unclassified LTRs: 10.67% in H. r. subsp. mongolica versus 22.82% in H. r. subsp. sinensis.
Based on homology evidence, transcript evidence and ab initio predictions, we annotated 56 217 protein-coding genes in H. r. subsp. mongolica and 57 160 in H. sinensis. In addition to these protein-coding genes, the H. r. subsp. mongolica assembly contains 791 rRNA, 1460 tRNA and 4897 ncRNA genes, whereas the H. r. subsp*. sinensis* assembly contains 1885 rRNA, 1200 tRNA and 5319 ncRNA genes. BUSCO assessment of the annotated protein-coding gene sets revealed completeness values of 99.2% for H. r. subsp. mongolica and 99.0% for H. r. subsp. sinensis. Functional annotation indicated that 98.05% of H. r. subsp. mongolica proteins and 98.15% of H. r. subsp. sinensis proteins could be assigned putative functions.
Each haplotype in our assemblies is approximately 1.5 times larger than the previously published chromosome-level assemblies of related subspecies (Fig. 1C). To explore the origin of these additional sequences, we conducted whole-genome comparisons using SyRI (V1.6) between the H. rhamnoides reference genome [4] and all haplotypes of H. r. subsp. mongolica and H. r. subsp. sinensis. The analyses revealed extensive structural rearrangements across all 12 chromosomes, dominated by frequent inter-chromosomal translocations and intra-chromosomal inversions (Fig. 1D). To further validate the authenticity of the structural inversions identified by SyRI, we examined the Hi-C interaction patterns between the two haplotypes of H. r. subsp. sinensis, which revealed off-diagonal contact signals consistent with the predicted inversion events (Fig. 1E). Compared with previously published assemblies [2, 4], each haplotype in our genomes contains substantially more sequences. To verify that these additional sequences, which were missing in earlier assemblies [4, 5], are genuine rather than artifacts of sequencing or assembly errors, we mapped publicly available HiFi reads of H. rhamnoides [4] and found no support for the expanded regions, whereas our own HiFi data provided full coverage, confirming that these sequences represent authentic genomic differences (Fig. 1F). More than 40% of aligned regions exhibited non-syntenic relationships, with inversions alone affecting 147–210 Mb of sequence in each haplotype comparison. In addition, non-syntenic or unaligned regions accounted for 28.3% to 48.4% of each query genome, highlighting substantial haplotype-specific variation, interspecies divergence, and the expansion of repetitive or structurally complex genomic regions. For instance, H. r. subsp. sinensis hapB contained 208 Mb of sequences absent from H. rhamnoides, while H. r. subsp. mongolica hapA carried more than 426 Mb of such regions (Fig. 1G).
To investigate the potential forces underlying these large-scale structural variations, we next examined the contribution of transposable elements (TEs). Genome-wide enrichment analysis revealed that LTR/Gypsy elements are non-randomly distributed, showing significant accumulation within structurally rearranged regions compared with randomized expectations (Fig. 1H). Notably, LTR/Gypsy elements were disproportionately enriched at chromosomal breakpoints (hapA: 2360; hapB: 1810) relative to broader inversion regions (hapA: 1547; hapB: 1308). These results suggest that LTR/Gypsy elements have actively mediated chromosome breakage and rearrangements, thereby playing a key role in shaping the distinct genome architectures of H. r. subsp. mongolica and H. r. subsp. sinensis.
The two subspecies exhibited notable differences in fruit pulp Vc content and seed oil content (Fig. 1I and J). H. r. subsp. sinensis accumulates significantly higher levels of Vc in the fruit pulp (Fig. 1I). To elucidate the molecular basis of this trait, we examined the ascorbate biosynthesis pathway. Several key genes, including GDP-l-galactose phosphorylase (VTC) (Hirsi04aG0052800), l-galactose dehydrogenase (l-GalDH) (Hirsi03aG0325000), and l-galactono 1,4-lactone dehydrogenase (GLDase) (Hirsi04aG0123700), exhibited markedly higher expression in H. r. subsp. sinensis relative to H. r. subsp. mongolica (Fig. 1K), consistent with the elevated Vc content. These genes exhibited statistically significant differential expression between the two species (t-test, P < 0.05), suggesting that their upregulation may contribute to the observed species-specific difference in Vc accumulation (Fig. 1I).
Conversely, H. r. subsp. mongolica exhibited significantly higher seed oil content than H. r. subsp. sinensis (Fig. 1I). Transcriptomic profiling of triacylglycerol (TAG) and fatty acid biosynthesis pathways revealed enhanced expression of an acyl-CoA:diacylglycerol acyltransferase (Hirmo06aG0056400), a key enzyme in TAG synthesis, and a species-specific glycerol-3-phosphate O-acyltransferase (GPAT) (Hirmo09aG0163800) in H. r. subsp.mongolica (Fig. 1L). In addition, two fatty aldehyde dehydrogenase (ALDH) (Hirmo07aG0056100 and Hirmo05aG0138100), along with a species-specific ALDH (Hirmo05aG0120300), were upregulated in the fatty acid pathway (Fig. 1M). Statistical tests confirmed that these genes are significantly more highly expressed in H. r. subsp. mongolica compared with H. r. subsp. sinensis (t-test, P < 0.05), supporting their contribution to the elevated seed oil content (Fig. 1J).
In summary, we present two gap-less, haplotype-resolved genomes of H. r. subsp. mongolica and H. r. subsp. sinensis, alongside tissue-specific transcriptomic data linked to key fruit quality traits. These resources not only provide a high-resolution view of genome architecture and gene content, but also offer mechanistic insights into the species-specific accumulation of Vc and seed oil. Together, they form a foundational platform for evolutionary studies and targeted breeding in seabuckthorn.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lan L, Leng L, Liu W. et al. The haplotype-resolved telomere-to-telomere carnation (Dianthus caryophyllus) genome reveals the correlation between genome architecture and gene expression. Hortic Res. 2023;11:uhad 24438225981 10.1093/hr/uhad 244PMC 10788775 · doi ↗ · pubmed ↗
- 2Yu L, Diao S, Zhang G. et al. Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides. Plant Biotechnol J. 2022;20:1257–7335244328 10.1111/pbi.13802 PMC 9241383 · doi ↗ · pubmed ↗
- 3Zhang G, Song Y, Chen N. et al. Chromosome-level genome assembly of Hippophae tibetana provides insights into high-altitude adaptation and flavonoid biosynthesis. BMC Biol. 2024;22:8238609969 10.1186/s 12915-024-01875-4PMC 11015584 · doi ↗ · pubmed ↗
- 4Wu Z, Chen H, Pan Y. et al. Genome of Hippophae rhamnoides provides insights into a conserved molecular mechanism in actinorhizal and rhizobial symbioses. New Phytol. 2022;235:276–9135118662 10.1111/nph.18017 · doi ↗ · pubmed ↗
- 5Yang X, Luo S, Yang S. et al. Chromosome-level genome assembly of Hippophae rhamnoides variety. Sci Data. 2024;11:77639003298 10.1038/s 41597-024-03549-w PMC 11246439 · doi ↗ · pubmed ↗