Late-Onset Huntington’s Disease: A Case Report and Literature Review
Carlos Gonçalves, Ana Sofia Ferreira, André Calheiros, Rafael Lopes Freitas, Goncalo Cacao

TL;DR
This paper reports a rare case of Huntington’s disease diagnosed in an 80-year-old man, emphasizing the importance of considering the disease in late-onset neurological symptoms.
Contribution
The novelty lies in presenting a confirmed case of late-onset Huntington’s disease without family history, aiding in diagnostic awareness.
Findings
An 80-year-old man with late-onset chorea and cognitive decline was diagnosed with Huntington’s disease via genetic testing.
Neuroimaging showed ischemic leukoencephalopathy, complicating the diagnosis of late-onset Huntington’s disease.
The case underscores the need to consider HD in differential diagnoses of late-onset neurological symptoms.
Abstract
Huntington’s disease (HD) is a rare autosomal dominant neurodegenerative disorder caused by expansion of the cytosine-adenine-guanine (CAG) trinucleotide repeat in the huntingtin (HTT) gene. Although the disease typically presents in mid-adulthood, symptom onset after the age of 60, defined as late-onset Huntington’s disease (LoHD), remains uncommon and may pose diagnostic challenges. We report the case of an 80-year-old man admitted for evaluation of progressive unintentional weight loss, whose clinical assessment revealed generalized chorea and progressive cognitive decline. Genetic testing identified an expanded CAG allele with 39 repeats, confirming the diagnosis of LoHD. Neuroimaging revealed ischemic leukoencephalopathy consistent with cerebral small vessel disease (CSVD), contributing to diagnostic complexity. This case highlights the importance of considering Huntington’s…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
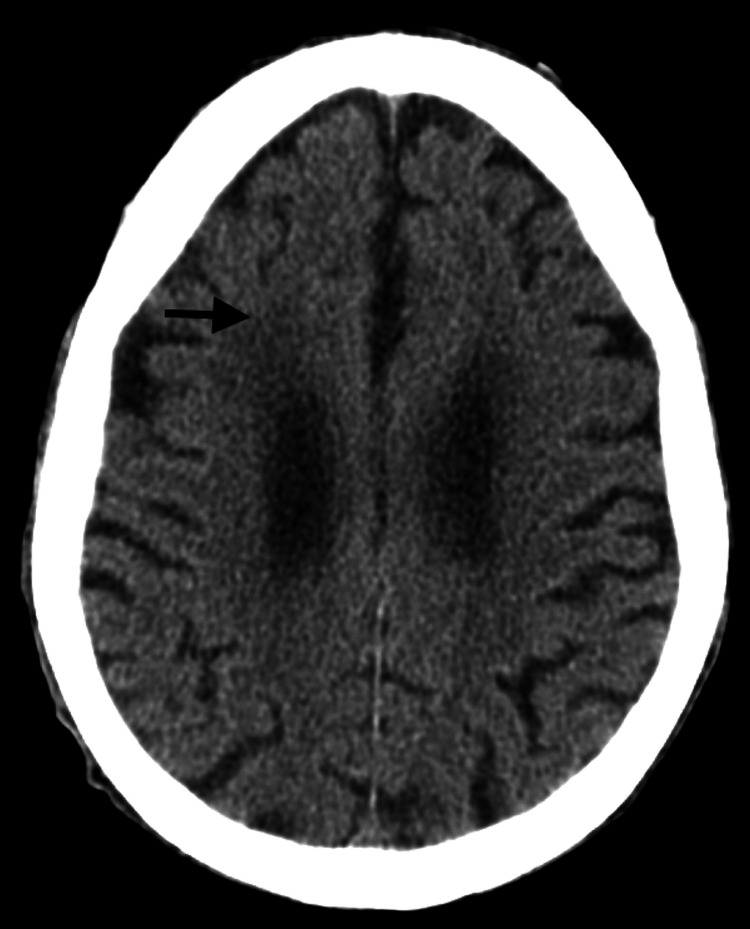 Figure 1
Figure 1| Laboratory test | Observed value | Normal range | |
| On admission | At discharge | ||
| Hemoglobin (g/dL) | 13.2 | 13.4 | 13.2-17.2 |
| Creatinine (mg/dL) | 3.35 | 0.61 | 0.60-1.30 |
| Urea (mg/dL) | 338 | 27 | 17-43 |
| Sodium (mmol/L) | 138 | 137 | 136-145 |
| Potassium (mmol/dL) | 3.9 | 5 | 3.5-5.1 |
| C-reactive protein (mg/dL) | <0.51 | <0.51 | <0.51 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Neurodegenerative Diseases · Amyotrophic Lateral Sclerosis Research · Cerebrovascular and genetic disorders
Introduction
Huntington’s disease (HD) is a rare autosomal dominant neurodegenerative disorder characterized by abnormal expansion of the cytosine-adenine-guanine (CAG) trinucleotide repeat in the huntingtin gene (HTT), leading to the production of a mutant huntingtin protein with toxic properties [1]. This results in synaptic dysfunction, transcriptional dysregulation, oxidative stress, and neuronal loss, predominantly affecting the striatum and giving rise to the classical clinical triad of motor, cognitive, and psychiatric manifestations [1]. The most common motor presentation is chorea, although rigidity, dystonia, and oculomotor abnormalities may also be observed [1].
The number of CAG repeats is the principal determinant of disease penetrance and age at onset. Alleles with ≥40 repeats are associated with complete penetrance and earlier disease onset, whereas alleles with 36-39 repeats demonstrate reduced penetrance and greater phenotypic variability [1]. Nevertheless, modifying factors, including deoxyribonucleic acid (DNA) repair genes such as MutS homolog 3 (MSH3) and Fanconi anemia-associated nuclease 1 (FAN1), significantly influence age at onset and disease progression, underscoring the complexity of the pathogenic mechanisms involved [2].
Late-onset Huntington’s disease (LoHD), defined as symptom onset after 60 years of age, has been reported to account for approximately 4.4%-11.5% of HD cases [3]. In this subgroup, the clinical phenotype is often more heterogeneous and frequently dominated by cognitive impairment or behavioral changes, while chorea may be less prominent. This contributes to diagnostic difficulty, particularly in differentiating LoHD from more prevalent neurodegenerative dementias and parkinsonian syndromes in older adults [2,4]. In older adults, clinical recognition may be further delayed by overlap with age-related neurodegenerative and vascular conditions (including cerebral small vessel disease (CSVD)), which may confound the interpretation of cognitive decline and functional deterioration [3,4]. Recent retrospective cohort data also highlight substantial diagnostic delay in LoHD, reinforcing the challenges of recognition in older adults [5]. Additionally, up to one-third of patients with LoHD do not report a clear family history, commonly due to reduced-penetrance alleles or unrecognized familial disease [4].
In advanced stages of HD, irrespective of age at onset, there is a transition toward a predominantly hypokinetic phenotype, characterized by rigidity, bradykinesia, and significant functional decline [1]. Cognitive deterioration progresses to a subcortical dementia with marked executive dysfunction and apathy. Nutritional complications, respiratory infections, and dysphagia become major contributors to morbidity and mortality [1]. In patients with LoHD, these features often overlap with age-related comorbidities, making a careful and multidisciplinary diagnostic approach essential [4].
Case presentation
An 80-year-old man with a history of arterial hypertension and no known family history of neurological or psychiatric disease was admitted for investigation of constitutional symptoms, including progressive unintentional weight loss. According to his family, involuntary movements had been present for approximately four years, initially mild and of small amplitude in the left foot, gradually progressing to involve all four limbs, the trunk, and the face, becoming increasingly pronounced. Over time, the patient developed progressive loss of autonomy in ambulation, personal hygiene, and feeding, concomitant with continued weight loss. In the months preceding admission, he also exhibited episodes of disorientation, behavioral changes, and fluctuations in attentional state.
On physical examination, the patient was markedly underweight, with generalized chorea and multidomain cognitive decline. Laboratory investigations (Table 1) revealed acute kidney injury of prerenal etiology, interpreted in the context of dehydration in a patient with reduced oral fluid and nutritional intake, without associated electrolyte disturbances. Cranial computed tomography (Figure 1) demonstrated ischemic leukoencephalopathy. No alternative cause for the weight loss was identified, and it was attributed to pronounced chorea associated with inadequate nutritional intake.
Non-contrast cranial computed tomography demonstrating ischemic leukoencephalopathy (arrow)
Given the suspicion of Huntington’s disease, a neurology consultation was requested, and a therapeutic trial with haloperidol was initiated, resulting in partial improvement of the involuntary movements. Genetic testing revealed one expanded CAG allele with 39 repeats and one normal allele with 11 repeats, confirming the diagnosis of Huntington’s disease. The patient remains under follow-up with symptomatic control achieved through neuroleptic therapy and restoration of adequate nutritional intake, and the family has been referred for genetic counseling.
Discussion
Late-onset Huntington’s disease (LoHD), typically defined as symptom onset after 60 years of age, remains an uncommon phenotype but is increasingly recognized. In older adults, diagnostic suspicion may be low, and clinical presentation can overlap with age-related neurological disorders, contributing to diagnostic delay. In a large retrospective cohort from Mexico, LoHD represented 7% of genetically confirmed Huntington’s disease cases, with a mean diagnostic delay of 6.2 years, emphasizing how frequently recognition is postponed in this subgroup [5]. In our patient, symptom onset in the eighth decade, together with prominent weight loss and progressive cognitive decline, contributed to diagnostic uncertainty and delayed identification.
Genotype-phenotype correlations are particularly relevant in LoHD. Expansions in the reduced penetrance range (36-39 CAG repeats) are associated with increased variability in clinical expression and later onset compared with fully penetrant expansions (≥40 repeats) [1,2]. The identification of 39 CAG repeats in this case aligns with the observation that LoHD often involves lower-range pathological expansions and may follow a more insidious disease evolution [3,4]. However, CAG repeat length alone does not fully account for age at onset, and multiple genetic and environmental modifiers influence disease penetrance and clinical course, including DNA repair pathways [2].
From a clinical standpoint, Huntington’s disease is characterized by progressive motor, cognitive, and psychiatric manifestations [1,6]. In older adults, cognitive decline and functional deterioration may dominate the clinical phenotype, while chorea may be initially subtle and attributed to other causes, further complicating recognition [4,5]. Psychiatric and behavioral symptoms also represent a major component of Huntington’s disease and may occur early or be misinterpreted as primary psychiatric illness, particularly in late-life presentations [7]. In addition, the absence of an apparent family history should not exclude Huntington’s disease. In LoHD cohorts, a substantial proportion of patients have unclear transmission patterns or no known affected relatives, likely due to reduced penetrance alleles, incomplete family history, small family structures, or unrecognized disease in relatives [4,5]. This was similarly observed in our case.
Neuroimaging interpretation in LoHD requires particular caution. While Huntington’s disease is classically associated with striatal involvement, older patients frequently demonstrate coexisting cerebrovascular pathology. In our patient, cranial computed tomography revealed ischemic leukoencephalopathy consistent with cerebral small vessel disease (CSVD). MRI was not available in this case to better assess striatal (caudate) atrophy. CSVD is common in advanced age and is independently associated with cognitive impairment, gait disturbance, and loss of functional autonomy. Therefore, CSVD likely contributed to the severity of cognitive decline and functional deterioration in this case and was an important confounder during the diagnostic process. Nevertheless, the progressive generalized chorea, cognitive decline, functional worsening, and molecular confirmation of HTT CAG expansion strongly support Huntington’s disease as the primary unifying diagnosis.
Accordingly, the differential diagnosis of late-onset chorea with cognitive decline should be broad. In elderly patients, clinicians should consider neurodegenerative conditions (e.g., dementia with Lewy bodies, frontotemporal dementia, and corticobasal syndrome), medication-induced hyperkinesias, autoimmune/paraneoplastic syndromes, metabolic causes, and structural lesions. In this clinical context, the frequent coexistence of age-related disorders such as CSVD reinforces the importance of systematic differential diagnosis and recognition that vascular pathology may coexist and amplify disability without fully explaining progressive chorea and a neurodegenerative trajectory [5,8].
Prognostically, LoHD should not be regarded as necessarily benign. In a multicenter South Korean study evaluating survival in LoHD, clinical and genetic variables were associated with survival outcome, and survival after onset remained limited, supporting the substantial morbidity burden in this population [8]. In our case, marked weight loss likely reflected a combination of hyperkinetic movements, reduced nutritional intake, and frailty, highlighting the importance of early nutritional evaluation and supportive interventions.
Although disease-modifying therapies are not yet available, the diagnosis has meaningful clinical implications. Definitive diagnosis requires molecular confirmation of HTT CAG repeat expansion [1,2]. Early genetic confirmation allows timely initiation of symptomatic treatment (e.g., neuroleptics for chorea), multidisciplinary supportive care, nutritional and swallowing assessment when appropriate, rehabilitation strategies to reduce falls, and referral for genetic counseling [1,7]. A limitation of this report is the lack of publication-ready MRI images to better illustrate caudate nucleus involvement and characterize concomitant cerebral small vessel disease.
Finally, emerging evidence suggests that Huntington’s disease may include a neurodevelopmental component that contributes to circuit vulnerability later in life, further underscoring the complex biology underlying clinical expression across the lifespan [9,10].
Conclusions
Late-onset Huntington’s disease is an uncommon but clinically important cause of progressive chorea and cognitive decline in older adults, even in the absence of a known family history. Given the frequent coexistence of age-related conditions such as cerebral small vessel disease, which may contribute to cognitive impairment and functional deterioration, careful differential diagnosis is essential. Definitive diagnosis requires molecular confirmation of HTT CAG repeat expansion, enabling appropriate symptomatic management, nutritional support, multidisciplinary care, and genetic counseling.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies Cold Spring Harb Perspect Med Jimenez-Sanchez M Licitra F Underwood BR Rubinsztein DC 7201710.1101/cshperspect.a 024240 PMC 549505527940602 · doi ↗ · pubmed ↗
- 2Huntington's disease: nearly four decades of human molecular genetics Hum Mol Genet Gusella JF Lee JM Mac Donald ME 06330202110.1093/hmg/ddab 170PMC 849001134169318 · doi ↗ · pubmed ↗
- 3What do we know about late onset Huntington’s disease?J Huntingtons Dis Chaganti SS Mc Cusker EA Loy CT 95103620172867113710.3233/JHD-170247 PMC 5502838 · doi ↗ · pubmed ↗
- 4Clinical and genetic characteristics of late-onset Huntington's disease in a large European cohort Eur J Neurol Petracca M Di Tella S Solito M 194019512920223535773610.1111/ene.15340 PMC 9324106 · doi ↗ · pubmed ↗
- 5Late-onset Huntington’s disease in Mexico: a retrospective study Cureus Ochoa-Morales A Beutelspacher-Fernandez K Jara-Prado A 017202510.7759/cureus.94482 PMC 1261000241234988 · doi ↗ · pubmed ↗
- 6From pathogenesis to therapeutics: a review of 150 years of Huntington’s disease research Int J Mol Sci Jiang A Handley RR Lehnert K Snell RG 24202310.3390/ijms 241613021 PMC 1045590037629202 · doi ↗ · pubmed ↗
- 7[Psychiatric symptoms of Huntington's disease]Nervenarzt Mühlbäck A Hoffmann R Pozzi NG Marziniak M Brieger P Dose M Priller J 8718849520243921268110.1007/s 00115-024-01728-z PMC 11374876 · doi ↗ · pubmed ↗
- 8Clinical and genetic characteristics associated with survival outcome in late-onset Huntington’s disease in South Korea J Clin Neurol Hwang YS Jo S Kim GH 3944012020243862722810.3988/jcn.2023.0329 PMC 11220345 · doi ↗ · pubmed ↗