High-Throughput Hyperspectral and Multiplexed Super-Resolution Fluorescence Imaging by SP-STORM
Elric Dion Pott, Meek Yang, James Ethan Batey, Joie Embree, Bin Dong

TL;DR
A new microscopy method called SP-STORM enables fast, high-resolution imaging of multiple subcellular structures simultaneously.
Contribution
SP-STORM introduces a novel approach combining spectral and spatial information for rapid, high-throughput super-resolution imaging.
Findings
SP-STORM simultaneously images five subcellular structures with minimal crosstalk.
The method is over ten times faster than existing multiplexing microscopy techniques.
SP-STORM is compatible with other super-resolution microscopy approaches.
Abstract
Simultaneous determination of spatial location and spectral color of single molecules at large molecular density with high throughput was achieved by combining single-molecule photoswitching and optical in-hardware Fourier transformation of single-molecule emission spectra into the phasor space. The method, named as spectral phasor enabled stochastic optical reconstruction microscopy (SP-STORM), achieved simultaneous super-resolution imaging of five subcellular structures in parallel with minimum crosstalk for the first time. The high-throughput feature of SP-STORM enables these subcellular structures to be readily resolved in about one min, which is more than an order of magnitude faster than other multiplexing single-molecule localization microscopy techniques. The concept of SP-STORM is also compatible with and can be readily applicable to other super-resolution microscopy.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
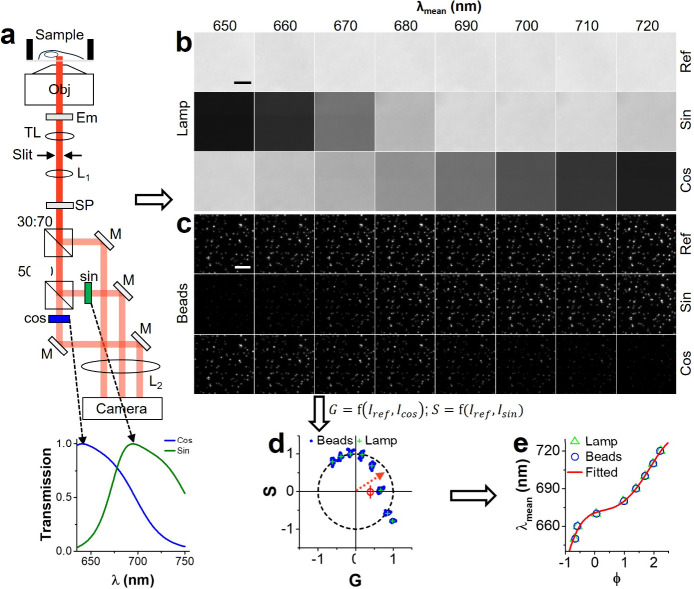 Figure 1
Figure 1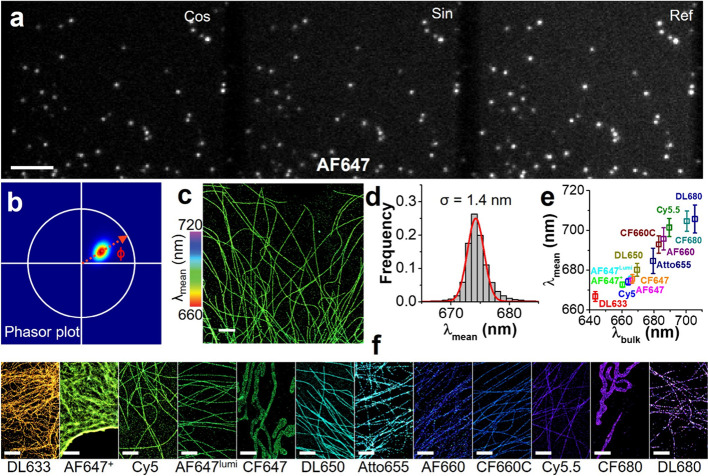 Figure 2
Figure 2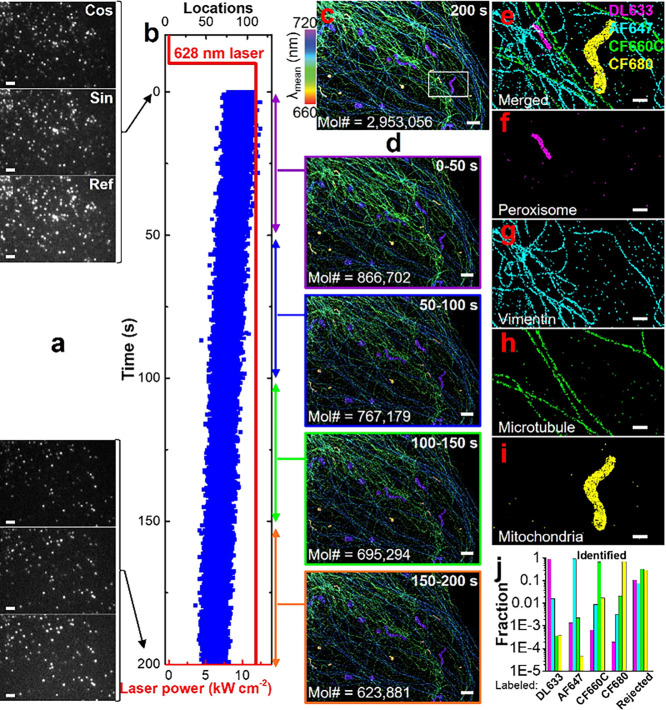 Figure 3
Figure 3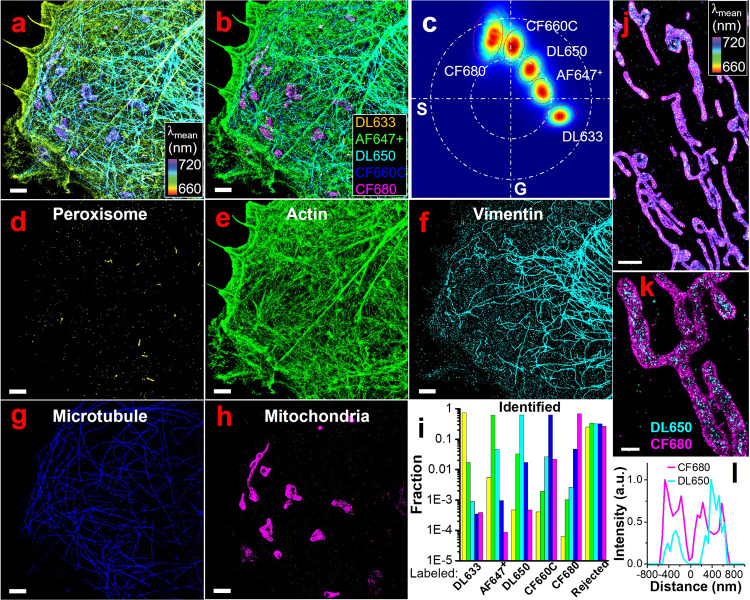 Figure 4
Figure 4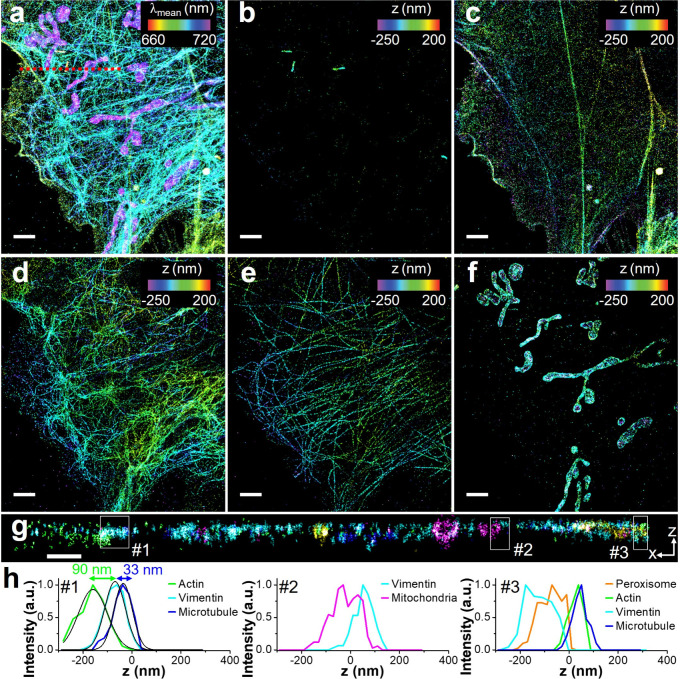 Figure 5
Figure 5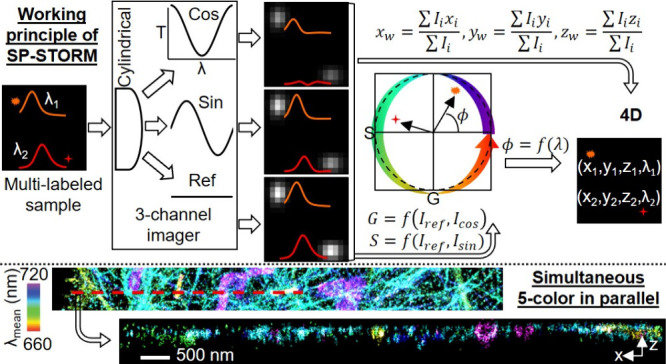 Figure 6
Figure 6 Figure 7
Figure 7- —Society for Analytical Chemists of Pittsburgh10.13039/100003413
- —University of Arkansas10.13039/100007756
- —Arkansas Biosciences Institute10.13039/100008231
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Fluorescence Microscopy Techniques · Digital Holography and Microscopy · Near-Field Optical Microscopy
Single-molecule localization-based super-resolved fluorescence microscopy (SMLM) ?,? has advanced studies of protein assemblies down to nanometer spatial resolution and even at single-protein resolution. ?,? Further advancements in multicolor SMLM attempt to explore its multiplexing capability. Utilizing spectrally well-separated fluorophores ?,? is the most straightforward approach, yet it requires long data acquisition time due to sequential image registration and involves complex error-prone alignment procedures between channels. A ratiometric-based spectral demixing approach enables simultaneously resolving up to four targets but suffers high crosstalk.? Dispersive (i.e., SR-STORM ?,? ) and excitation modulation (i.e., ExR-STORM?) based spectral demixing approaches can simultaneously resolve four targets with low crosstalk. However, both suffer low throughput due to either a low molecular density requirement to avoid spectral interference (SR-STORM) or mechanical switching between excitation laser lines (ExR-STORM). Four-color SR-STORM and ExR-STORM take 12 and 25 min, respectively. In comparison, single-color dSTORM requires less than a minute. ?,?
DNA-PAINT is another type of multiplexed SMLM with high spatial resolution.? It has theoretically unlimited multiplexing capability in combination with solution exchange (Exchange-PAINT).? However, the throughput of this approach remains a major limitation. Recent optimizations, such as FRET-based probes? and fluorogenic DNA-PAINT,? have improved acquisition speed at the expense of reducing multiplexing capacity. The development of secondary label-based unlimited multiplexed PAINT (SUM-PAINT)? attempted to address this trade-off by decoupling the DNA barcoding of the target from the imaging process. The time to quantitatively map up to 30 proteins in neuron cells at single-protein resolution was reduced from 800 h using Exchange-PAINT? to 30 h using SUM-PAINT. Nevertheless, the intrinsic nature of sequential imaging in SUM-PAINT remains as the bottleneck for high-throughput analysis.
Here, we developed a nondispersive approach to simultaneously determine single-molecule locations and their fluorescence emission spectra with high throughput. The approach is based on the integration of single-molecule imaging and optical in-hardware Fourier transformation of single molecules’ emission spectra into the phasor space for spectral demixing (i.e., in-hardware spectral phasor analysis). In this work, we integrate the in-hardware spectral phasor analysis with dSTORM? imaging, named as SP-STORM. We simultaneously obtained multiplexed super-resolution microscopy image of 5 protein targets in parallel and with minimum crosstalk in ∼1 min. This is more than an order of magnitude faster than all previously developed multiplexed SMLM techniques which can simultaneously resolve up to 4 protein targets in few tens of minutes to hours. We further demonstrated SP-STORM’s compatibility for 3D super-resolution imaging.
In SP-STORM, we modulated single molecules’ fluorescence emission using a lab-built three-channel imager (Figurea, Figure S1), where the signals in two of the channels were transformed by cosine- and sine-shaped optical bandpass filters (Figurea, inset), namely, the sine and cosine channels. The unmodified signal in the remaining channel works as the reference for phasor analysis. Using the photon number of the same single molecules in three channels, one can obtain their locations in the phasor space (Figure, S2) where the phase angle (ϕ) is wavelength-dependent. We calibrated the imaging system to establish the spectral mean-phase angle relationship by refining the spectral color of light from a microscope built-in lamp or emission from fluorescence beads using narrow bandpass filters (Figureb-d). The obtained spectral mean-phase angle relationship follows a nonlinear trend that was well-fit by a fourth-order polynomial function (Figuree). This calibration curve was then used to determine the spectral mean of single molecules in dSTORM imaging experiments. Notably, optical filters with transmission profiles representing complete sine and cosine functions are ideal. ?,? However, optical filters with broadband transmission similar to ideal sine/cosine filters can achieve the same purpose with a modified analysis procedure (Figure S3).? Nevertheless, we speculated that using nonideal transmission optical filters could result in larger errors in the obtained spectral information. Using simulation data, we evaluated such impact, and the results suggest that the error from using nonideal optical filters is minimum (Additional discussion in Figure S4).
We first tested the widely used dSTORM dye Alexa fluor 647 (AF647) for SP-STORM imaging. We labeled the fixed cells through immunostaining and then photoswitched most of AF647 dye molecules into a nonfluorescent dark state (i.e., off state) resulting in only a subset of dye molecules fluorescing (i.e., on state) at any given instant. By optimizing the excitation laser power density (>10 kW cm^–2^) and imaging acquisition rate (200 Hz), the transformed (i.e., cosine and sine) and unmodified (i.e., reference) emission signals from sparsely distributed single AF647 molecules were simultaneously recorded at different regions of the same camera (Figurea). The same single AF647 molecules in three channels were localized and grouped. Their photon numbers were measured and used to determine single AF647 molecules’ phase angles in the phasor space (Figureb), which enables the quantification of single-molecule spectral means using the established calibration curve (Figuree). The targeted subcellular structures were then reconstructed using the weighted spatial locations of single AF647 molecules in three channels with colors representing the single-molecule spectral mean (Figurec). The localization precision for AF647 was estimated to be ∼ 6 nm from small cluster analysis (Figure S7), which corresponds to a spatial resolution of ∼ 15 nm (full width at half-maximum) comparable to typical dSTORM imaging. The histogram distribution of >10^5^ single AF647 molecules (Figured) gives an average spectral mean of 674.2 nm and a spectral variation of 1.4 nm (s.d.). Such small spectral variation holds promise for highly multiplexed SMLM by spectral demixing dyes with minimal spectral differences.
However, significant broadening and fluctuation of spectral color at single molecule level has been well-known due to factors including photon noise, background noise, sensitivity of the detector, and inherent heterogeneity. It has been reported that single molecules can show a spectral shift of up to 30 nm in emission peak. ?,? We evaluated another 12 far-red organic dyes (λ_bulk_: 644–706 nm, Figure S5–S17). Representative SP-STORM images are shown in Figuref. The spatial resolution ranges from 15 to 40 nm. The results of the single-molecule spectral mean and variation for each dye are shown in Figuree. Dye molecules with bulk emission peak differences as small as 5 nm (e.g., AF647 vs DL650) can be readily resolved by SP-STORM due to their small single-molecule spectral variations. Therefore, theoretically, approximately 23 dyes can be spectrally resolved by SP-STORM in the spectral window of 635–750 nm, assuming all dyes own such small single-molecule spectral variation and can be efficiently excited and photoswitched by a single 628 nm laser. However, the results show a spectral variation ranging from 1.3 to 7.1 nm for all tested far-red dyes, with a general trend of smaller single-molecule spectral variation for brighter dyes (Figure S18). Furthermore, most of the tested dyes show a single population of spectral color, except AF660 and DL680 (Figures S13, S17), highlighting the importance of assessing the inherent heterogeneity of dye molecules at the single molecule level. Importantly, these screening results laid the foundation for multiplexed SMLM by choosing dyes based on photoswitching capability, spatial resolution, spectral variation, and overlapping in the phasor space.
To test SP-STORM for multiplexed imaging and compare its throughput to other methods, we labeled four distinct subcellular targets in fixed cells with four dyes of heavily overlapped emission spectra. A single 628 nm laser was used for exciting and photoswitching all dye molecules between on and off states (Figurea). Excitingly, the targeted four subcellular structures are readily distinguishable based on the spectral mean alone (Figurec). Distinct colors of purple, blue, green, and yellow were clearly observed for labeled mitochondria, microtubule, vimentin, and peroxisome, respectively. Single molecules were further classified for multiplexed images by comparing their locations in the phasor space to that of known dye molecules (Figure S19g). dSTORM images of labeled subcellular structures show negligible misidentification (Figuree-i, Figure S19b-f) with color crosstalk <2% (Figurej). We then evaluated the throughput of SP-STORM. The number of locations per image frame along with the data acquisition time was plotted in Figureb. The average number of localizations per image frame was estimated to be ∼ 74, corresponding a density of ∼ 0.14 locations per μm^2^. Under this density of single molecules, multiplexed dSTORM images with highly resolvable subcellular structures are already readily available in less than a minute (Figured) based on the Nyquist limit? (Figure S20).
We further tested the feasibility of SP-STORM for simultaneous super-resolution imaging of five targets (Figure). Five subcellular structures are readily observed with distinct colors (Figurea), where purple, blue, cyan, yellow-green, and yellow denote mitochondria, microtubule, vimentin, actin, and peroxisome, correspondingly. The multiplexed dSTORM image was readily obtained in ∼ 1.3 min (Figure S21). For all channels, the crosstalk was determined to be <5% (Figureb-i). Simultaneous 5-color SMLM imaging in parallel has never been achieved previously. To evaluate if the superlocalized positions align well for different dye molecules, we performed SP-STORM imaging of mitochondrial outer membrane protein (i.e., TOM20) and mitochondrial heat shock protein (i.e., mtHsp70 or Mortalin, primarily localizes inside the mitochondria) with CF680 and DL650. The hyperspectral and multiplexed dSTORM images (Figurej,k) and the cross-section profile (Figurel) clearly match the expectation that locations of DL650 are fully surrounded by CF680.
We next demonstrated the capability of SP-STORM for 3D super-resolution imaging. Here, we adapted astigmatism-based 3D SMLM technique? by introducing a weak cylindrical lens (1000 mm focal length) at approximately 2 cm away from the intermediate image plane (Figures S1, S22). By combining the spectral and spatial information, a 4D (i.e., x, y, z, and λ) super-resolution image was obtained (Figure). The low color crosstalk remains, where negligible misidentification between five dye channels was observed (Figureb-f). Notably, here 3D dSTORM images were presented in individual dye channels. The 3D localization precision of sub-10 nm (lateral) and sub-20 nm (axial) were achieved for all dyes (Figure S23), similar to previously reported results.? Figureg shows a cross-section image in a xz plane, providing a more complete visualization of spatial arrangement and interaction of targeted subcellular structures. We selected three regions for cross-section profile analysis of the distributions of five super-resolved structures as examples: the nanoscale layer-by-layer distribution of cytoskeleton filaments at site #1, the contact of microtubules on the top of a mitochondrion at site #2, and the insertion of peroxisome between layers of cytoskeleton filaments at site #3.
The key challenges in advancing techniques for quantifying biomolecules and their spatial locations include sensitivity, spatial resolution, multiplexing capability, and throughput. Previously, the advancement of SMLM techniques enabled the simultaneous measurement of up to four protein targets in parallel or sequential quantification of theoretically unlimited protein targets with nanometer spatial resolution. However, the intrinsic nature of these techniques, either low SNR, low molecular density, low data acquisition rate, or sequential data acquisition, limits their throughput. The unique strength of SP-STORM lies in its advantages of a nondispersive approach and using cosine- and sine-shaped broadband optical filters. It enables high-density single-molecule imaging with high SNR resulting in a low spectral uncertainty of measurement, which ultimately leads to the high throughput and large multiplexing capability of SP-STORM. SP-STORM has demonstrated the capacity to simultaneously resolve five distinct protein targets in parallel and with low crosstalk in about a minute, which is more than an order of magnitude faster than all current multiplexed SMLM techniques (i.e., a few tens of minutes to hours for four protein targets). The advantages and limitations of current multiplexed SMLM techniques including SP-STORM were summarized in Table S1.
While it is beyond the scope of the current work, SP-STORM is readily adaptable for other applications, such as studying the transformation of intermediates or isomers in chemical reactions where distinct emission profiles are present, and sensing local environment at nanoscale using environmental sensitive dyes (e.g., solvatochromic dye, Nile red). Furthermore, the high-throughput feature of SP-STORM also holds promises for studying dynamic processes in live cells. By applying high laser power (>10 kW cm^–2^) and fast data acquisition speed (∼kHz), single-color dSTORM imaging of live cell membrane dynamics was achieved with 1–2 s temporal resolution.? However, one needs to carefully assess the potential phototoxicity when operating under such a high illumination intensity. Additionally, motion blur due to sample drift can potentially introduce artifacts into the results of cellular dynamic processes. One can add nonphotobleaching fiducial markers (e.g., gold nanoparticles) to either lock the sample in three-dimensional or correct drift in post data analysis. Lastly, the integration of spectral phasor analysis for spectral demixing of fluorophores is broadly compatible with other super-resolution techniques, e.g., (F) PALM, PAINT, and STED, highlighting its versatility and potential for advancing multiplexed microscopy across various imaging platforms. However, it is worth noting that applying spectral phasor analysis for microscopy imaging with bulk emission? (e.g., STED) potentially risks high color crosstalk for dyes with heavily overlapped spectra.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Betzig E.Patterson G. H.Sougrat R.Lindwasser O. W.Olenych S.Bonifacino J. S.Davidson M. W.Lippincott-Schwartz J.Hess H. F.Imaging Intracellular Fluorescent Proteins at Nanometer Resolution Science 200631357931642164510.1126/science.112734416902090 · doi ↗ · pubmed ↗
- 2Rust M. J.Bates M.Zhuang X.Sub-Diffraction-Limit Imaging by Stochastic Optical Reconstruction Microscopy (STORM)Nat. Methods 200631079379610.1038/nmeth 92916896339 PMC 2700296 · doi ↗ · pubmed ↗
- 3Balzarotti F.Eilers Y.Gwosch K. C.GynnåA. H.Westphal V.Stefani F. D.Elf J.Hell S. W.Nanometer resolution imaging and tracking of fluorescent molecules with minimal photon fluxes Science 2017355632560610.1126/science.aak 991328008086 · doi ↗ · pubmed ↗
- 4Schmidt R.Weihs T.Wurm C. A.Jansen I.Rehman J.Sahl S. J.Hell S. W.MINFLUX nanometer-scale 3D imaging and microsecond-range tracking on a common fluorescence microscope Nat. Commun.2021121147810.1038/s 41467-021-21652-z 33674570 PMC 7935904 · doi ↗ · pubmed ↗
- 5Bates M.Huang B.Dempsey G. T.Zhuang X.Multicolor Super-Resolution Imaging with Photo-Switchable Fluorescent Probes Science 20073175845174910.1126/science.114659817702910 PMC 2633025 · doi ↗ · pubmed ↗
- 6Dempsey G. T.Vaughan J. C.Chen K. H.Bates M.Zhuang X.Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging Nat. Methods 20118121027103610.1038/nmeth.176822056676 PMC 3272503 · doi ↗ · pubmed ↗
- 7Testa I.Wurm C. A.Medda R.Rothermel E.von Middendorf C.Fölling J.Jakobs S.Schönle A.Hell S. W.Eggeling C.Multicolor Fluorescence Nanoscopy in Fixed and Living Cells by Exciting Conventional Fluorophores with a Single Wavelength Biophys. J.20109982686269410.1016/j.bpj.2010.08.01220959110 PMC 2956215 · doi ↗ · pubmed ↗
- 8Zhang Z.Kenny S. J.Hauser M.Li W.Xu K.Ultrahigh-throughput single-molecule spectroscopy and spectrally resolved super-resolution microscopy Nat. Methods 20151293510.1038/nmeth.352826280329 · doi ↗ · pubmed ↗