Pyracylenes: Facile Synthetic Access and Continuous Face-To-Face Antiaromatic π‑Stacking
Sheng-Yuan Cheng, Jiun-Siang Juang, Yu-Fang Huang, Ting-Yi Lai, Yi-Hung Liu, Fumitaka Ishiwari, Akinori Saeki, Kenji Okada, Ryohei Kishi, Jeffrey M. Farrell

TL;DR
This paper presents a new method to synthesize pyracylenes and reveals their unique antiaromatic π-stacking behavior in the solid state.
Contribution
A facile Pd-catalyzed synthesis of pyracylenes and the discovery of continuous face-to-face antiaromatic π-stacking in the solid state.
Findings
Pyracylenes were synthesized via Pd-catalyzed ring contraction of diborinic acids.
Three pyracylenes formed continuous face-to-face antiaromatic π-stacks with equidistant spacing and 90° twist angles.
NICS calculations showed reduced antiaromaticity in the stacked pyracylene structures.
Abstract
Pyracylene is a nonalternant hydrocarbon which has long fascinated chemists, but whose structure is exceedingly rare in experimental reports. Pyracylene’s surprising absence arises from synthetic inaccessibility attributable to its antiaromatic character. Meanwhile, sterically unimpeded polycyclic antiaromatic hydrocarbons have become fascinating candidates for organic materials applications and for antiaromatic π-stacked assemblies. Herein, we report a facile synthesis of pyracylenes via Pd-catalyzed ring contraction of diborinic acids. So-formed pyracylenes were derivatized by bromination and subsequent Pd-catalyzed cross-coupling. All pyracylenes were isolated, fully characterized, and studied by UV–vis absorption spectroscopy, cyclic voltammetry, density functional theory (DFT) calculations, and X-ray crystallography. In the solid state, three pyracylenes formed unprecedented…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1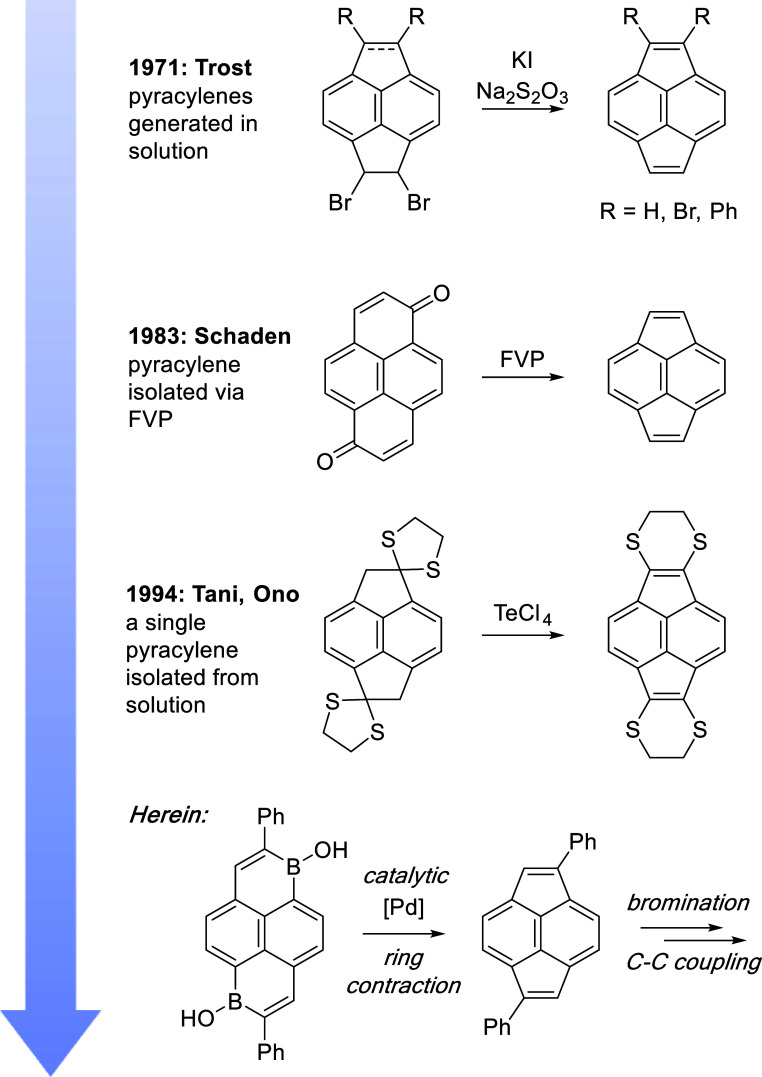 2
2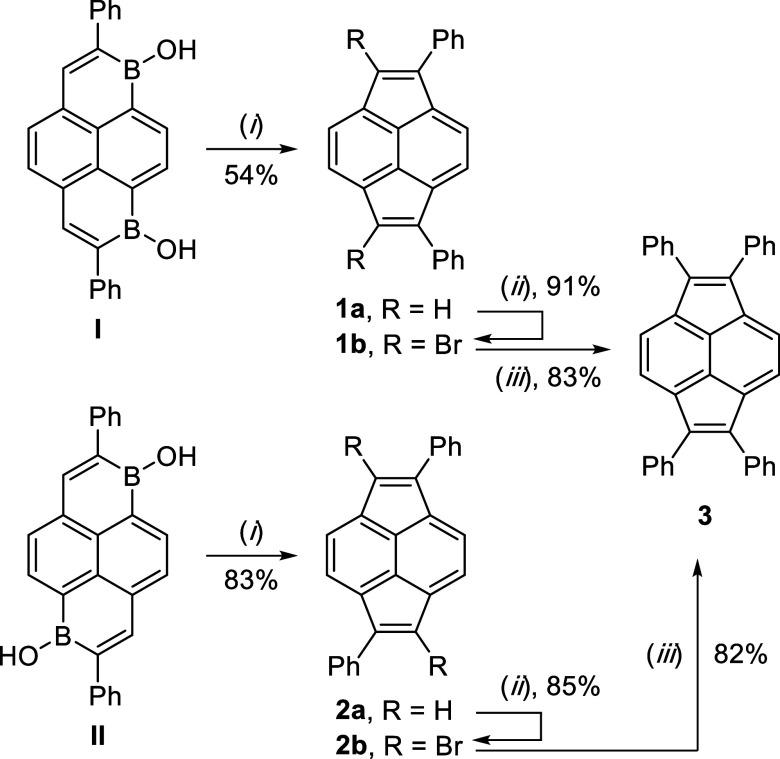 1
1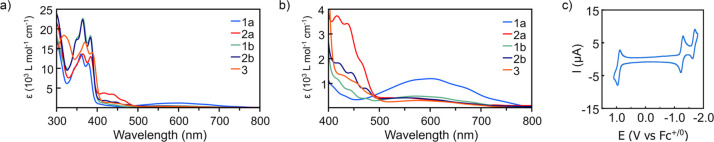 3
3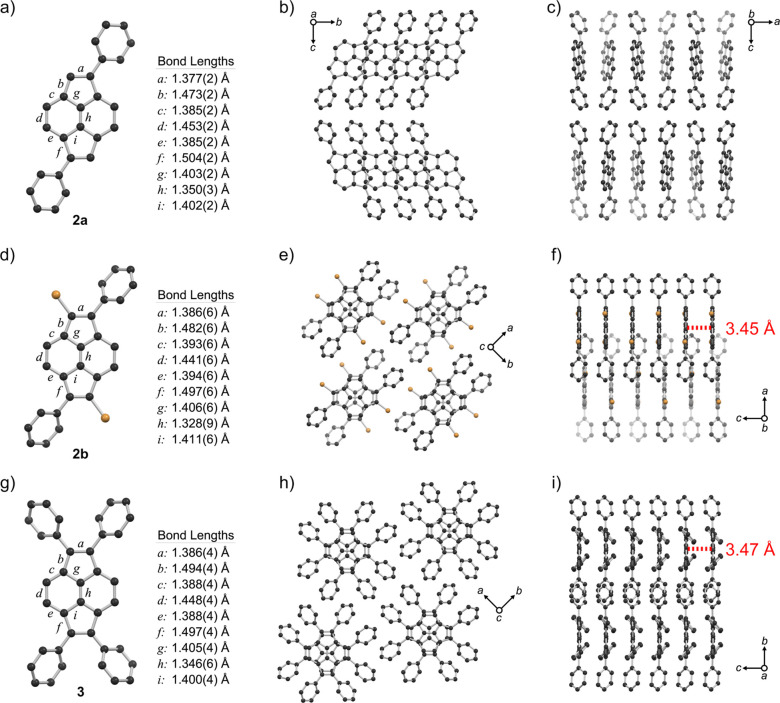 4
4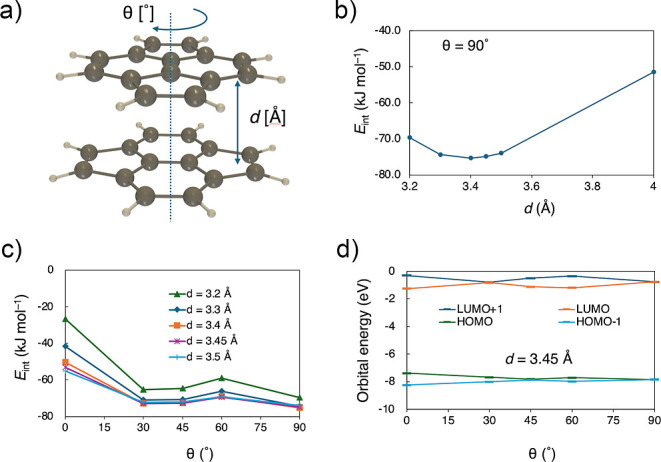 5
5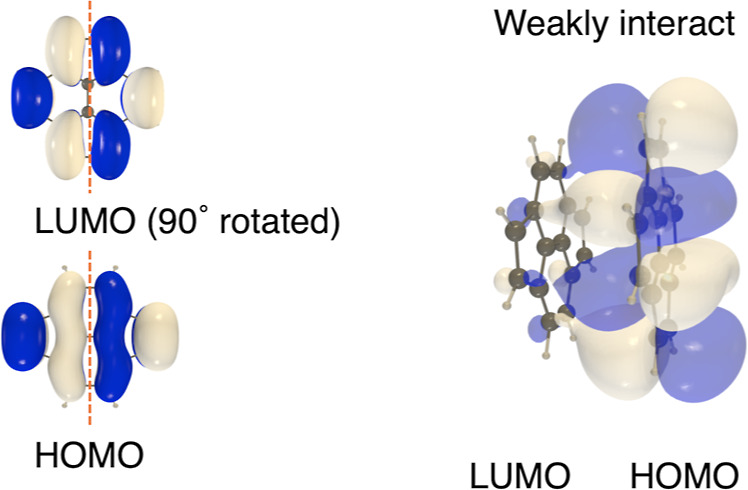 6
6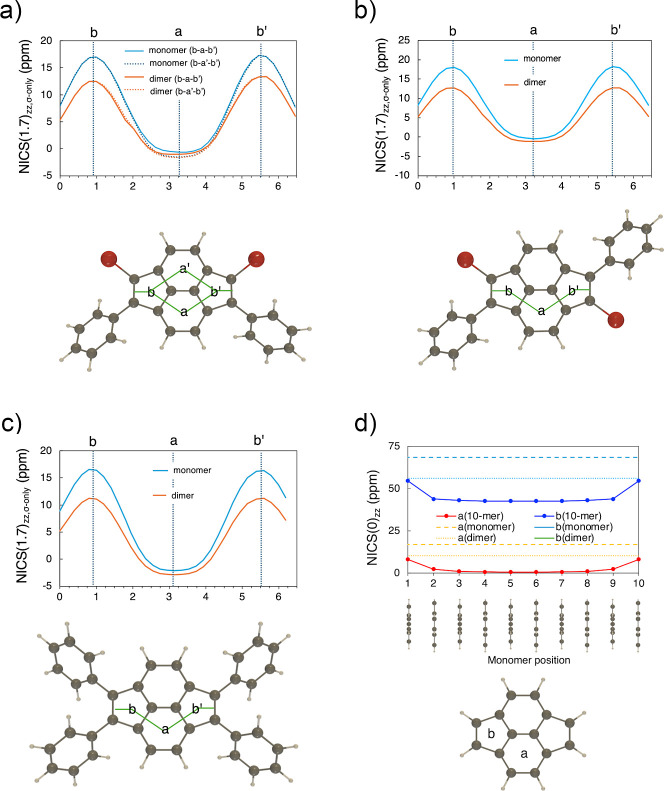 7
7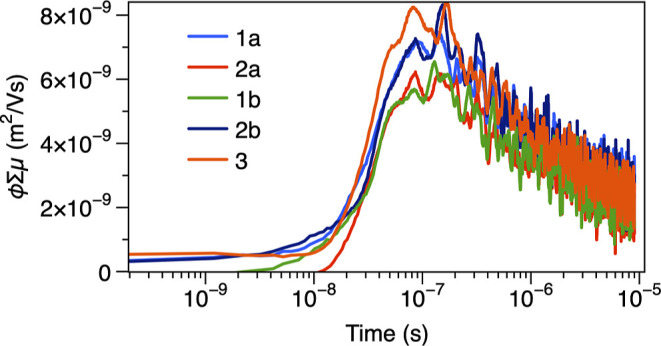 8
8| λabs [nm] (ε [M–1 cm–1]) |
|
|
|
| |
|---|---|---|---|---|---|
|
| 600 (1200), 377 (11,100), 360 (13,600) | 0.63 | –1.47 (−1.50) | - (−1.90) | 2.06 |
|
| 514 (400), 436 (3400), 417 (3700), 384 (13,000), 365 (13,600) | 0.66 | –1.47 (−1.48) | - (−1.87) | 2.10 |
|
| 572 (500), 383 (18,300), 365 (22,700) | 0.92 | –1.26 | –1.67 | 2.18 |
|
| 561 (400), 470 (800), 444 (1400), 383 (17,800), 364 (22,300) | 0.91 | –1.28 | –1.67 | 2.19 |
|
| 578 (300), 387 (14,000), 370 (16,600), 318 (18,400) | 0.67 | –1.45 (−1.44) | - (−1.80) | 2.12 |
- —Japan Society for the Promotion of Science10.13039/501100001691
- —Japan Society for the Promotion of Science10.13039/501100001691
- —National Taiwan University10.13039/501100006477
- —National Science and Technology Council10.13039/501100020950
- —National Science and Technology Council10.13039/501100020950
- —Ministry of Education, TaiwanNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Properties of Aromatic Compounds · Synthesis and pharmacology of benzodiazepine derivatives · Catalytic Cross-Coupling Reactions
Introduction
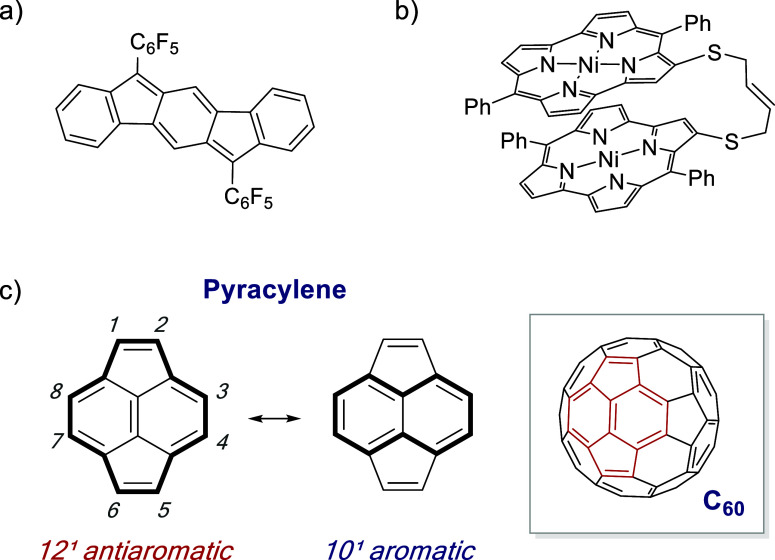
Polycyclic aromatic hydrocarbons (PAHs) are pervasive structures throughout chemistry and materials science. Polycyclic antiaromatic hydrocarbons (PAAHs) are comparatively less explored due to characteristic high reactivities that lead to problematic syntheses and handling, but have experienced growing interest. Unique properties of PAAHs, such as small HOMO–LUMO energy gaps, single-molecule conductivities,? unique excited-state dynamics,? and relationships to diradicals,? could potentially produce transformative functional organic materials.? Indeed, small HOMO–LUMO energy gaps and charge-carrier transport properties of some PAAHs (e.g., Figurea) have been leveraged for organic transistors.?
(a) A PAAH used in transistors by Haley, Nuckolls, and co-workers. (b) Example of a face-to-face π-stacked norcorrole dimer reported by Shinokubo, Kim, Kowalczyk, and co-workers. (c) The structure of pyracylene illustrating (anti)aromatic circuits and its relationship to fullerene (inset).
The unique antiaromatic electronic structures of PAAH π-systems also suggest fundamentally different π–π interactions than those between PAH π-systems. For example, face-to-face π-stacking of antiaromatic π-systems may diminish or eliminate their antiaromaticities.? Shinokubo and co-workers have recently reported breakthrough experimental evidence for this “stacked-ring aromaticity” in face-to-face π-stacked norcorrole oligomers (e.g., Figureb).? However, to our knowledge there have been no reports of related antiaromaticity-stabilizing orbital interactions in continuous, well-defined, face-to-face π-stacks. Indeed, there have been relatively few experimental studies of π-stacked assemblies of PAAHs in general, which has limited understanding of their unique interactions and self-assembly behaviors. Contributing to experimental difficulties, PAAH stabilization via sterics is largely precluded for such studies, as this impedes π–π stacking.
Pyracylene (Figurec) is a planar nonalternant hydrocarbon (and fullerene substructure) which can be described as a 10π-electron [4n + 2] aromatic naphthalene system bearing two ethylene bridges, or as a 12π-electron [4n] antiaromatic system based on its conjugated periphery.? Although pyracylene has intrigued chemists for decades,? this deceptively simple molecule and its derivatives remain nearly absent from experimental accounts. After a failed synthetic attempt by Anderson and Anderson,? seminal work from Trost and co-workers reported the generation of pyracylenes in solution (Figure). ?,? These species eluded isolation, but exhibited comparatively upfield-shifted periphery ^1^H chemical shifts attributable to the effects of paramagnetic ring currents. ?,?,? Pyracylene was finally isolated via flash vacuum pyrolysis by Schaden in low yield, ?,? and studies of this molecule by the groups of Wirz? and Minas Da Piedade? indicated that the molecule is energetically stable and readily handled in pure form, despite antiaromatic character on magnetic and electronic grounds. This suggests (1) that pyracylenes bear a combination of stability and antiaromatic character, and (2) that the current dearth of pyracylenes is a problem of synthetic methodology rather than inherent instability. While fusion of aromatic rings at the pyracylene 1,2 and 5,6 positions allows synthetic access to species with fascinating properties,? these are magnetically and electronically distinct from pyracylene, and often lack antiaromatic characteristics. To our knowledge, the lone example of a nonfused pyracylene isolated from a solution-phase synthesis is a 1,2:5,6-bis(ethylenedithio)pyracylene synthesized via tellurium tetrachloride mediated ring expansion (Figure).?
Timeline of progress in the synthesis of pyracylenes and the work presented herein.
Herein we describe a facile, catalytic, solution-phase synthesis of ambient-stable pyracylenes via Pd-catalyzed ring contraction of polycyclic borinic acids. These pyracylenes were readily brominated in high yields, and were subsequently amenable to Suzuki–Miyaura cross-coupling. All pyracylenes herein were studied by ^1^H and ^13^C NMR spectroscopy, high-resolution mass spectrometry (HRMS), UV–vis absorption spectroscopy, cyclic voltammetry, density functional theory (DFT) calculations, and X-ray crystallography. In some cases, the solid-state structures of pyracylenes revealed striking 1D π-stacks with equidistant molecular spacings, wherein adjacent molecules adopted unusual face-to-face positionings with 90° twist angles. DFT calculations indicated that this stacking arrangement aligned HOMOs and LUMOs of adjacent pyracylene monomers and diminished their antiaromaticities. Time-resolved microwave conductivity (TRMC) measurements of crystalline pyracylenes revealed conductivities in the solid state.
Results and Discsussion
Synthesis of Pyracylenes 1–3
We envisioned Pd-catalyzed ring-contraction of previously reported 2,7-diphenyl-1,8-dihydroxy-1,8-diborapyrene (I) or 2,7-diphenyl-1,6-dihydroxy-1,6-diborapyrene (II) as potential routes to pyracylenes (Figure and Scheme). Precursors I and II are conveniently prepared using a one-pot procedure of Würthner and co-workers.? Moreover, Fu and co-workers have previously reported Pd-catalyzed ring contractions of dibenzoxaborininols? that intimate possible pyracylene synthesis by ring contractions of I or II.
Optimized Catalytic Synthesis of Pyracylenes 1a and 2a via Ring Contraction, Synthesis of Brominated Pyracylenes 1b and 2b, and Suzuki Coupling Reactions of 1b and 2b to Form 3
To explore a ring contraction route to a pyracylene, we probed 1:2 stoichiometric reactions of I with Pd(OAc)2 in CD_3_CN. Heating this reaction at 70 °C for 30 min gave rise to distinctive ^1^H NMR singlets consistent with paratropically shifted pyracylene periphery protons (δ = 6.47, 6.76, and 7.18) that we attributed to structure 1a. An analogous reaction was carried out on a preparative scale in CH_3_CN at 70 °C, from which pyracylene 1a could be isolated as a green solid in 11% yield after solvent removal in vacuo and purification by column chromatography. The structure of pyracylene 1a was confirmed by ^1^H and ^13^C NMR spectroscopy as well as HRMS (see Supporting Information). Repeating our stoichiometric NMR-scale reactions of I with Pd(PPh_3_)4, PdCl_2_(PPh_3_)2, Ni(OAc)2, or Cu(OAc)2 in place of Pd(OAc)2 led to no observable pyracylene formation by ^1^H NMR spectroscopy.
We then sought to develop a catalytic variant of the ring contraction of I. While Fu and co-workers’ Pd-catalyzed ring contraction of dibenzoxaborininols involved oxidative cleavage of a B–C bond by I_2_ prior to Suzuki-type coupling,? the C–C bond-forming step of Pd-mediated ring-contraction of I apparently requires no exogenous oxidant, indicating a divergent mechanism. Nevertheless, an oxidant would presumably be required to regenerate an active Pd species for a catalytic cycle. Thus, we began catalytic screening by reacting I with 20 mol % Pd(OAc)2 in CH_3_CN at 70 °C for 2 h with 2 equiv of 1,4-benzoquinone as a terminal oxidant (see Supporting Information Table S1). Pyracylene 1a could be isolated from this reaction in 14% yield, indicating probable catalytic turnover. Use of water as a cosolvent (1:10 water/CH_3_CN) led to a lower yield for this reaction, but when used in conjunction with 10 equiv of [n-Bu_4_N][Br], isolated yields of 1a increased to 35%. Performing the reaction under N_2_ atmosphere further increased the yield to 54%, although degassing solvent offered no further improvement. Replacing CH_3_CN solvent with toluene offered similar yields of 1a (56%, or 54% upon scale-up), whereas replacement with DMF, DMSO, or dioxane led to inferior yields. Using otherwise optimized conditions, replacement of 1,4-benzoquinone with Cu(OAc)2, AgOAc, TEMPO, or MnO_2_ gave inferior yields. Our optimized catalytic conditions were then applied to the reaction of precursor II. Thus, II was reacted in the presence of Pd(OAc)2 (20 mol %), 1,4-Benzoquinone (2 equiv), and [n-Bu_4_N][Br] (10 equiv) at 70 °C for 2 h in acetonitrile/water. In this case, product isolation was facilitated by the poor solubility of 2a in this solvent mixture. Pure 2a was isolated as a dark green solid in 83% yield after solvent washing steps, filtration of a CH_2_Cl_2_ solution through Celite, and solvent removal in vacuo.
To explore possible avenues of derivatization, 1a and 2a were each reacted with two equivalents of N-bromosuccinimide in CHCl_3_ for 3 h (−35 °Cr.t.) to afford dibrominated pyracylenes 1b and 2b in high yields (Scheme). In contrast, in situ reactions of pyracylenes with Br_2_ performed by Trost and co-workers led to addition products rather than bromide-substituted pyracylenes.? Brominated pyracylenes 1b and 2b were then probed for Pd-catalyzed C–C coupling reactivity as a possible derivatization route. To our delight, 1b and 2b were each effective coupling partners in Suzuki–Miyaura reactions with phenyl boronic acid, catalyzed by 10 mol % Pd(PPh_3_)4 in toluene for 16 h at 100 °C with Cs_2_CO_3_ as a base (Scheme). In each reaction, a red precipitate formed that could be isolated by filtration, washing, and recrystallization from hot CHCl_3_ to yield pure 1,2,5,6-tetraphenylpyracylene 3 in 83% or 82% yield from 1b or 2b, respectively.
Pyracylenes 1–3 were fully characterized by ^1^H NMR spectroscopy, ^13^C NMR spectroscopy, HRMS, and single-crystal X-ray crystallography. For each, relatively upfield ^1^H chemical shifts were observed for protons on the pyracylene periphery compared to structurally related 1-phenylacenaphthylene, 1,2-dihydropyracylene, or 1,2-diphenyl-5,6-dihydropyracylene (see Supporting Information). ?,? Echoing previous reports of the stability of pyracylene in pure form,? solid samples of 1–3 stored in air for 7 days showed no observable decomposition by ^1^H NMR spectroscopy. Solution-phase stabilities were assessed in CDCl_3_ solutions kept under ambient, aerobic conditions. While 1a shows onset of decomposition after 1 day by ^1^H NMR spectroscopy, 2a shows onset of decomposition after 2 days. Remarkably, brominated pyracylenes 1b and 2b showed no signs of decomposition after 7 days in solution. The tendency of 3 to precipitate from solution over time prevented accurate assessment of its solution-phase stability. Sublimations of each compound 1–3 were possible under vacuum (10^–5^ Torr) at temperatures between 110 °C–160 °C (see Supporting Information for details). Interestingly, Jenneskens and co-workers have noted that unsubstituted pyracylene could not be resublimed after FVP synthesis.?
UV–Vis Spectroscopy
and Voltammetry Studies of 1–3
UV–vis measurements were performed on ∼10^–5^ M solutions of pyracylenes 1–3 in CH_2_Cl_2_ (Figurea,b and Table). Notably, each compound exhibited a broad absorption band, featuring low absorption intensity in the region of ∼500–800 nm. Such weak long-wavelength absorptions are typical for PAAHs, representing their low-energy, but symmetry-forbidden, HOMO–LUMO transitions.? The UV–vis spectra of 1–3 simulated by TD-DFT calculations were in good agreement with the experimental results (Figure S55). The reason for the relatively large absorption intensity in the region of ∼500–800 nm in 1a is due to a larger symmetry-lowering effect of the substituent introduction (see Supporting Information). Fluorescence spectroscopy indicated no measurable emissions from dilute (∼10^–5^ M) solutions of 1–3 in degassed CH_2_Cl_2_ under excitation at various λ_ex_ (each close to respective local absorption maxima of 1–3, see Supporting Information). The nonemissive behavior of these compounds in solution is consistent with unsubstituted pyracylene,? and is common for antiaromatic molecules in general.
(a) UV–vis absorption spectra of pyracylenes 1–3 (∼10–5 M in CH2Cl2, 298 K). (b) Magnified 400–800 nm regions of UV–vis absorption spectra of pyracylenes 1–3 (∼10–5 M in CH2Cl2, 298 K). (c) Cyclic voltammogram of 1b (1.6 × 10–4 M, 0.1 M [n-Bu4N][PF6], in CH2Cl2, vs Fc+/0, 298 K).
1: Optical and Electronic Properties of 1–3
Redox properties of compounds 1–3 were studied by cyclic voltammetry in 10^–4^ to 10^–3^ M solutions in 0.1 M [n-Bu_4_N][PF_6_] CH_2_Cl_2_ (e.g., Figurec and Table). Facile, reversible one-electron reductions were measured for each compound. These occur at −1.45 V to −1.47 V vs Fc^+/0^ for 1a, 2a, and 3, reflecting their electron-deficient fused five-membered ring-containing π-systems. These also fall in line with unsubstituted pyracylene, for which a first reduction at −1.056 V vs SCE in DMF was reported. For brominated 1b and 2b, first reversible one-electron reductions occur at −1.26 V and −1.28 V vs Fc^+/0^, approximately 0.2 V more positive than 1a, 2a, or 3. For comparison the first one-electron reduction potential of popular fullerene derivative PC_61_BM is reported at −1.17 V vs Fc^+/0^ (in o-dichlorobenzene).? In addition, second overall quasi-reversible one-electron reductions were observed for 1b and 2b within the electrochemical window of CH_2_Cl_2_, both at −1.67 V vs Fc^+/0^. Using THF as a solvent allowed observation of second, irreversible one-electron reductions for 1a, 2a, and 3 with peak maxima at −1.90 V, −1.87 V, and −1.80 V vs Fc^+/0^, respectively. Redox amphoterism of PAAHs has implications on ambipolar charge-carrier transport. Notably, quasi-reversible one-electron oxidations were measured for 1b, 2b, and 3 in CH_2_Cl_2_ at 0.92, 0.91, and 0.67 V vs Fc^+/0^, respectively. Irreversible oxidation events were observed for 1a and 2a, with peak potentials at 0.63 and 0.66 V, respectively. From voltammetric data, HOMO–LUMO energy gaps ranging 2.06–2.19 eV were estimated for 1–3 (Table).
Crystallographic Studies
Pyracylenes 1a–b, 2a–b, and 3 were studied by X-ray crystallography (Figures and S53). The pyracylene cores of each of these compounds are planar with only slight deviations. For example, 1a shows a slight curvature with planes defined by 5-membered rings deviating ∼8.6° from coplanarity (Figure S50), and 3 shows a torsion angle of ∼3.5° about the central CC bond (Figure S51). In all cases, significant bond-length alternations are observed between carbons of the pyracylene cores, consistent with the reported solid-state structure of unsubstituted pyracylene.? Thus, perimeter C–C bonds of 1–3 alternate between shorter lengths <1.41 Å (e.g., Figurea,d,g, bonds labeled a, c and e), and longer lengths >1.44 Å (e.g., Figurea,d,g, bonds labeled b, d and f). The central C–C bonds of 1–3 (e.g., Figurea,d,g, bonds labeled h) are short, with bond lengths resembling C–C double bonds (1.32 Å–1.35 Å). The optimized structures from DFT calculations agree well with experimental results in terms of both bond lengths and patterns of bond alternation, although the central CC bond lengths of 1b and 2b were about 0.01 Å longer than the experimental results (Figure S54).
Solid state structures of pyracylenes 2a, 2b, and 3 highlighting molecular structures and bond lengths (a: 2a, d: 2b, g: 3), as well as solid-state packing arrangements (b,c: 2a, e,f: 2b, h,i: 3). C: black, Br: orange, H atoms omitted for clarity.
Strikingly, pyracylenes 1b, 2b, and 3 arrange into continuous 1D π-stacks with unusual packings of polycyclic antiaromatic cores, exhibiting 90° twist angles and negligible parallel displacements between adjacent molecules (Figurese,f,h,i, and S53e,f). Equal spacings between pyracylene cores were calculated with centroid–mean plane distances of 3.41 Å (1b), 3.45 Å (2b), and 3.47 Å (3). This structural characteristic differs from 1a and 2a, which do not form π-stacks. The unique, face-to-face 1D stacking of 1b, 2b, and 3 also differs from previously reported structures of pyracylenes. ?,?,? The packings of 1b, 2b, and 3 contrast with the usual parallel-displaced π-stacks observed for aromatic molecules, where displacement mitigates repulsive interactions. Indeed, packings of 1b, 2b, and 3 can be compared to (+), or “Greek cross,” aggregate structures of aromatic π-systems, for which very few examples have been crystallographically characterized.? In solid-state structures, close CH-π distances (<∼3.0 Å) were observed between orthogonally oriented phenyl substituents on neighboring pyracylene molecules (Figure S52). These occur between adjacent molecules within 1D π-stacks of 1b, 2b, and 3, and additionally between molecules in adjacent stacks of 1b and 3.
Analysis of Aromaticity and Stacking Interactions
The intermolecular interactions in the 90°-twisted stacking system were analyzed using DFT calculations. The dimeric part was extracted from the solid-state structure of 2b, and the energy decomposition analysis (EDA) for the intermolecular interaction energy was conducted (see Supporting Information). The absolutely localized MO (ALMO-)EDA? indicated that the dispersion term was the main contributor in the dimer of 2b. On the other hand, the contribution of charge-transfer (CT) interactions related to orbital interactions were not significant due to the stacking distance of 3.45 Å, where orbital overlap is limited.
Next, we constructed a face-to-face dimer model based on the optimized structure of unsubstituted pyracylene and examined the potential energy surface for the stacking distance d and twist angle θ (Figurea). The results indicated a local minimum around θ = 90° and d ∼ 3.4 Å (Figureb,c), resembling the orientations of 1b, 2b, and 3 in the solid state. In this orientation, the orbital interactions between the HOMOs (LUMOs) of the pyracylene dimer vanish (Figured), suggesting that the destabilization of the HOMO caused by π-stacking is minimal, while dispersion forces provide significant stabilization. This is an interesting divergence from typical aromatic π-stacks, wherein parallel displacements mitigate repulsions.
Structure of unsubstituted pyracylene dimer model (a), d-dependence of the interaction energy E int for models with θ = 90° (b), θ-dependence of E int (c), and θ-dependence of the orbital energies for models with d = 3.45 Å (d), calculated at the ωB97M-V/def2-SVPD level.
Although the orbital interaction between the HOMOs (LUMOs) of monomers vanishes at θ = 90°, that between the HOMO of one monomer and the LUMO of the other monomer does not vanish. Interestingly, at this angle, the phase of the HOMO of one molecule matches that of the LUMO of the other molecule (Figure). Although this effect appears to be small at the stacking distance observed in the solid-state structures of 1b, 2b, and 3 (d ∼ 3.45 Å), to our knowledge there are no reports of this type of orbital interaction mode in one-dimensional π-stacks of antiaromatic molecules. Our calculations indicate that this HOMO–LUMO orbital interaction between pyracylene monomers should become important at shorter stacking distances (Figure S61), suggesting future design considerations for stabilization of antiaromatic π-stacked structures. For example, molecular designs of pyracylene which reduce HOMO–LUMO gaps may strengthen such orbital interactions and reduce stacking distances. Here it is worth contrasting with aromatic compounds, which are typified by HOMOs and LUMOs of different symmetry where the LUMO is bisected by an additional nodal plane compared to the HOMO. Thus, due to phase considerations, the face-to-face HOMO–LUMO interaction of two identical aromatic π-surfaces may be expected to be net nonbonding at any rotation of neighboring molecules.
Diagrammatic representation of the orbital interaction between the HOMO of one pyracylene monomer and the LUMO of another pyracylene monomer at θ = 90° and d = 3.45 Å.
Magnetic response properties were evaluated at the GIAO-RB3LYP/6-311++G(d,p) level (see Supporting Information). The geometries were taken from the solid-state structure and XY-scans of NICS_πzz,σ‑only_ values were evaluated 1.7 Å above the molecular plane (Figures and S58).? Consistent with previous calculations of unsubstituted pyracyclene,? positive NICS_πzz,σ‑only_ values indicated paratropicity primarily localized on five-membered rings of 1–3.
Results of NICS-XY scan of the monomer and π-stacked dimer of 1b (a), 2b (b), and 3 (c), with geometries based on solid-state structures, and (d) NICS(0) zz of the 10-mer model consisting of unsubstituted pyracylenes with d = 3.45 Å and θ = 90°, calculated at the GIAO-RB3LYP/6-311++G(d,p) level.
We then assessed the effect of intermolecular interactions on NICS_πzz,σ‑only_ values of 1b, 2b, and 3 by using dimer models with geometries extracted from solid-state structures. For the dimers, the NICS(1.7)πzz,σ‑only was scanned outside the dimer structure (Figurea–c). The NICS(1.7)πzz,σ‑only values of the five-membered rings in the dimeric structures were diminished compared to those in the monomer, indicating that intermolecular interactions between pyracylenes in this stacking arrangement could reduce the paratropicity of five-membered rings to some extent and increase the diatropicity of six-membered rings slightly.
Nevertheless, 1b, 2b, and 3 are shown to form infinite one-dimensional π-stacking structures with uniform stacking distances (d) in the solid state. To explore the effect of such continuous, face-to-face, 90°-twist-angle stacking arrangements on (anti)aromaticity, NICS calculations were performed for a 10-mer model consisting of unsubstituted pyracylenes with d = 3.45 Å and θ = 90°. NICS(0)_ zz _ values were evaluated at the centers of the five- and six-membered rings of each monomer (Figured). The NICS(0)_ zz _ values in the middle region of the 10-mer were almost converged.
The converged value on the five-membered ring (∼42.6 ppm) was significantly decreased from the NICS(0)_ zz _ value of the monomer (68.5 ppm), and even smaller than that of a similarly assessed dimer model (56.0 ppm). These results suggest that antiaromatic character also weakens in the continuous stacking case of pyracylene with face-to-face 90°-twist-angle arrangements of monomers.
The reduction of antiaromatic character caused by molecular stacking has been previously discussed using a mechanism involving orbital level switching due to strong interactions between the HOMOs (LUMOs) for fully face-to-face (θ ∼ 0° and d ∼ 3 Å) dimers of cyclobutadiene and Ni(II) norcorrole. ?,? Intriguingly, paratropicity reduction in face-to-face, 90°-twist-angle arrangements of pyracylenes, with orthogonally oriented HOMOs (LUMOs) and vanishing HOMO–HOMO (LUMO–LUMO) interactions, evidently does not follow this mechanism. For the pyracylenes computationally examined herein, we propose that the interactions of the HOMO of one monomer and the LUMO of another monomer, although weak at d ∼ 3.45 Å, influence magnetic response properties. Such interactions are expected to increase the HOMO–LUMO gap (Δε_HL_ = ε_LUMO_ – ε_HOMO_) in the dimer (or n-mer) compared to that of the monomer (Figure S60). Noting the molecular orbital contribution to magnetic responses (see Supporting Information), these changes to Δε_HL_ can in turn be expected to affect NICS values. Indeed, the NICS_ zz _ values for five-membered rings of unsubstituted pyracylene monomers and 90°-twisted π-stacking dimers at d = 3.45 Å and 10.0 Å show correspondence to Δε_HL_ (Table S7). Furthermore, a canonical MO-based decomposition analysis of the NICS (CMO-NICS)? enabled us to ascribe the trend of total NICS_ zz _ to trends in the contribution from the degenerate HOMO and HOMO–1. These results suggest that the intermolecular HOMO–LUMO orbital interactions between 90°-twisted π-stacked pyracylenes lead to an increase of Δε_HL_ which in turn reduces paratropicities (see Supporting Information, Table S7).
Conductivity Measurements
Motivated by emerging applications of PAAHs in electronic materials,? we measured time-resolved microwave conductivity (TRMC) profiles? of crystalline samples of 1–3 (Figure). Crystalline samples of 1b, 2b, and 3, which possess 1D π-stacking channels, exhibited clear TRMC signals with φΣμ ≈ 7 × 10^–9^ m^2^/(V s) comparable to those of typical π-conjugated polymers.? To compare with other antiaromatic compounds, while one specific derivative in antiaromatic norcorrole liquid crystals was reported to exhibit a relatively high φΣμ_max_ value of up to 5.2 × 10^–8^ m^2^/(V s),? most reported antiaromatic systems show φΣμ_max_ values on the order of ∼0.1–1 × 10^–9^ m^2^/(V s).? The photoconductivities observed in our systems are comparable in magnitude to these typical values. Interestingly, no orbital interactions between the HOMOs (LUMOs) of adjacent monomers of unsubstituted pyracylene are expected in 90°-twisted stacking arrangements. Nevertheless, observed conductivities of 1b, 2b, and 3 may be explained by substituent-induced symmetry reduction of frontier molecular orbitals that allows electronic coupling between neighboring stacked molecules (see Supporting Information), or by conducting pathways other than the stacking direction. Crystals of 1a and 2a, which possess herringbone arrangements of pyracylene π-surfaces in the solid state, also exhibited TRMC signals comparable to those of 1b, 2b, and 3.
TRMC profiles of 1a (blue), 2a (red), 1b (green), 2b (deep blue), and 3 (orange).
Conclusions
An expedient solution-phase synthesis of pyracylenes was achieved through Pd-catalyzed ring contraction of diborinic acids. The resulting pyracylenes underwent further derivatization via bromination and Pd-catalyzed cross-coupling reactions. All pyracylene products were fully characterized by ^1^H and ^13^C NMR spectroscopy, high-resolution mass spectrometry, and single-crystal X-ray crystallography. The electronic, optical, and magnetic properties of these products were studied using UV–vis absorption spectroscopy, cyclic voltammetry, and DFT calculations. Despite characteristic antiaromatic properties revealed by these studies, the pyracylenes were stable under ambient conditions.
Three pyracylene derivatives synthesized herein assembled into face-to-face one-dimensional antiaromatic π-stacks in the solid state, featuring uniform intermolecular distances and 90° twist angles between adjacent monomers. DFT calculations indicated that this stacking arrangement aligns HOMOs of antiaromatic monomers with phase-complementary LUMOs of neighboring molecules (and vice versa) while diminishing antiaromaticities of individual pyracylene molecules. Time-resolved microwave conductivity measurements revealed conductivities for pyracylenes in the solid state.
These results provide renewed access to a fundamental and intriguing nonalternant hydrocarbon scaffold, and have revealed unique characteristics of pyracylene that we hope will further understanding of antiaromatic π-stacking and present new prospects for antiaromatic materials design.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Chen W.Li H.Widawsky J. R.Appayee C.Venkataraman L.Breslow R.Aromaticity Decreases Single-Molecule Junction Conductance J. Am. Chem. Soc.201413691892010.1021/ja 411143 s 24397414 · doi ↗ · pubmed ↗
- 2a Rosenberg M.Dahlstrand C.KilsåK.Ottosson H.Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations Chem. Rev.20141145379542510.1021/cr 300471 v 24712859 · doi ↗ · pubmed ↗
- 3a Abe M.Diradicals Chem. Rev.20131137011708810.1021/cr 400056 a 23883325 · doi ↗ · pubmed ↗
- 4a Hong C.Baltazar J.Tovar J. D.Manifestations of Antiaromaticity in Organic Materials: Case Studies of Cyclobutadiene, Borole, and Pentalene Eur. J. Org. Chem.20222022 e 20210134310.1002/ejoc.202101343 · doi ↗
- 5a Kawase T.Fujiwara T.Kitamura C.Konishi A.Hirao Y.Matsumoto K.Kurata H.Kubo T.Shinamura S.Mori H.Miyazaki E.Takimiya K.Dinaphthopentalenes: Pentalene Derivatives for Organic Thin-Film Transistors Angew. Chem., Int. Ed.2010497728773210.1002/anie.20100360920836110 · doi ↗ · pubmed ↗
- 6a Corminboeuf C.von Ragué Schleyer P.Warner P.Are Antiaromatic Rings Stacked Face-to-Face Aromatic?Org. Lett.200793263326610.1021/ol 071183 y 17658752 · doi ↗ · pubmed ↗
- 7a Nozawa R.Tanaka H.Cha W.-Y.Hong Y.Hisaki I.Shimizu S.Shin J.-Y.Kowalczyk T.Irle S.Kim D.Shinokubo H.Stacked antiaromatic porphyrins Nat. Commun.201671362010.1038/ncomms 1362027901014 PMC 5141365 · doi ↗ · pubmed ↗
- 8a Anderson A. G.Anderson R. G.Attempts to Prepare Pyracylene. 1,2-Dihydropyracylene J. Org. Chem.19582351752010.1021/jo 01098 a 003 · doi ↗