Benzochromanone and Benzochromene Natural Products: Synthetic Strategies and Total Syntheses
Takuya Kumamoto

TL;DR
This paper presents new synthetic methods to create natural products with benzochromanone and benzochromene structures, enabling the production of bioactive compounds like xanthones and blennolide C.
Contribution
The paper introduces versatile and enantioselective synthetic strategies for benzochromanone and benzochromene natural products.
Findings
Total syntheses of (±)-4-deoxyblennolide C, (+)-blennolide C, and gonytolide C were achieved.
An asymmetric synthesis of (R)-(+)-teretifolione B and methylteretifolione B was successfully developed.
A Diels–Alder reaction using benzyne and oxygenated furans enabled efficient benzochromene synthesis.
Abstract
This account overviews our synthetic strategies for natural products featuring benzochromanone and benzochromene frameworks. The total synthesis of monomeric benzochromanones, particularly xanthones and benzochromanones is achieved. Key accomplishments include the development of a versatile synthetic approach for constructing xanthone frameworks via spirochromanone intermediates and the successful total syntheses of (±)‐4‐deoxyblennolide C, (+)‐blennolide C, and chromanone lactone gonytolide C. The asymmetric total synthesis of benzochromene (R)‐(+)‐teretifolione B and the first asymmetric synthesis of (R)‐(+)‐methylteretifolione B are also achieved. The Diels–Alder reaction between benzyne derived from chromene precursors and oxygenated furans enabled efficient access to benzochromene derivatives. The enantioselective synthesis of teretifolione B and related compound was accomplished…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
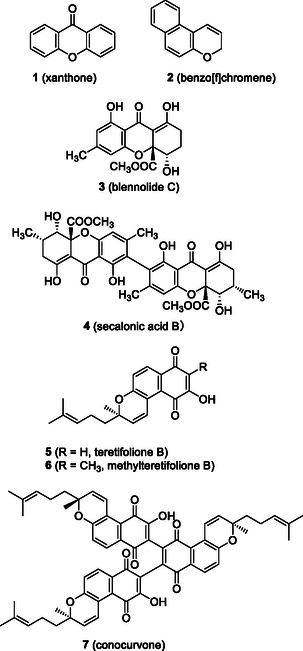 FIGURE 1
FIGURE 1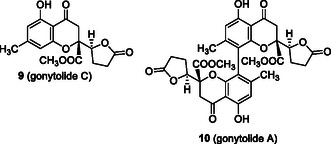 FIGURE 2
FIGURE 2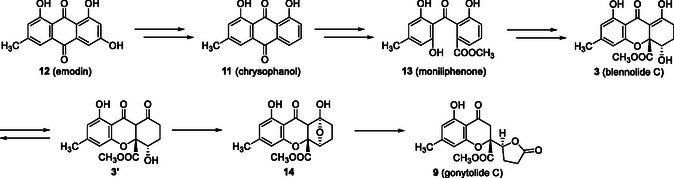 SCHEME 1
SCHEME 1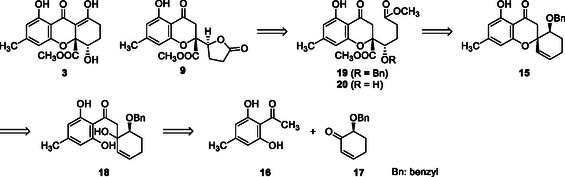 SCHEME 2
SCHEME 2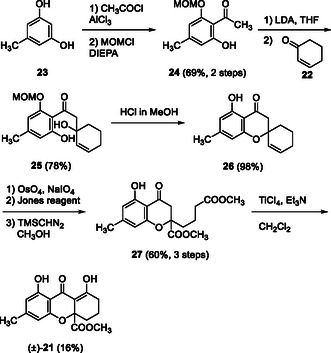 SCHEME 3
SCHEME 3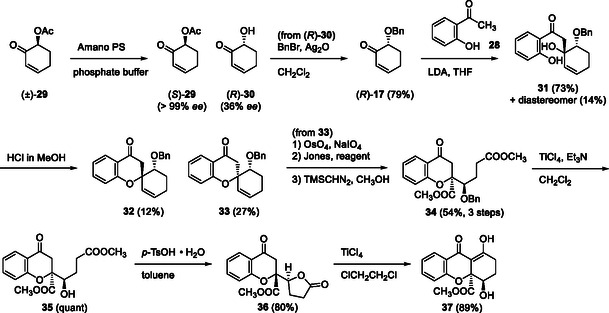 SCHEME 4
SCHEME 4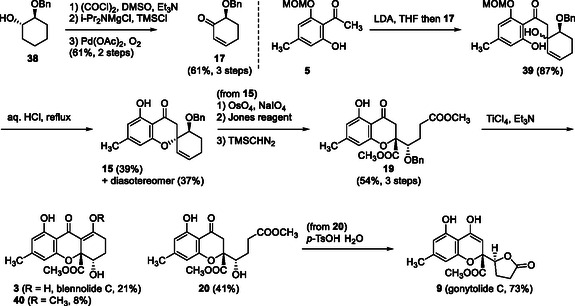 SCHEME 5
SCHEME 5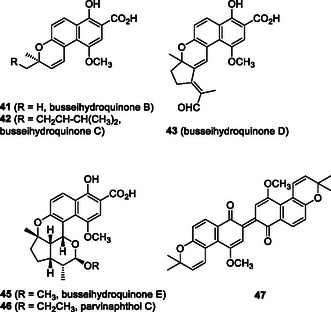 FIGURE 3
FIGURE 3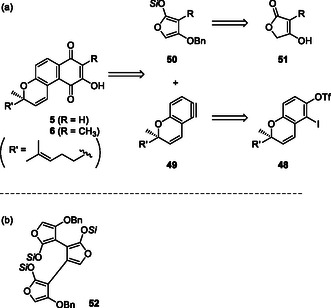 SCHEME 6
SCHEME 6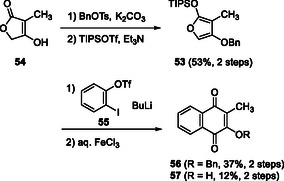 SCHEME 7
SCHEME 7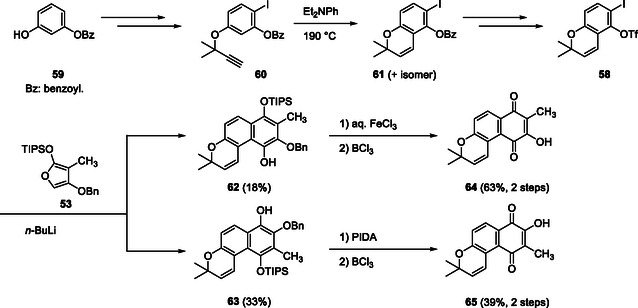 SCHEME 8
SCHEME 8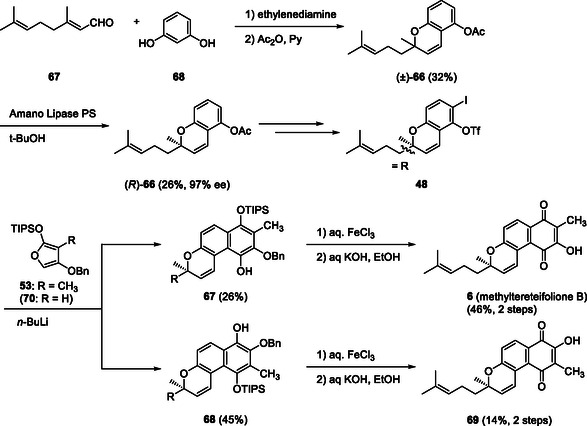 SCHEME 9
SCHEME 9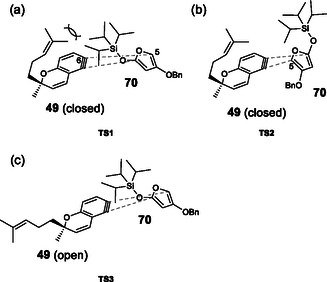 FIGURE 4
FIGURE 4|
| |||
|---|---|---|---|
| Run | Solvent | Conditions |
|
| 1 | THF | −78°C, 20 min | 1 : 1.7 |
| 2 | THF | −40°C, 20 min | 1 : 1.8 |
| 3 | THF | 0°C, 20 min | 1 : 1.9 |
| 4 | DME | −78°C, 30 min | 1: 1.7 |
| 5 | Hexane | −78°C, 30 min | 1 : 3.3 |
| 6 | Toluene | −78°C, 30 min | 1 : 3.2 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNatural Compound Pharmacology Studies · Synthesis of Organic Compounds · Cyclization and Aryne Chemistry
Introduction
1
Natural products containing benzochromanone and benzochromene structures have been isolated from a wide range of natural sources, including plants, actinomycetes, lichens, and endophytes, and are found in both monomeric and oligomeric forms (Figure 1) [1, 2]. Our research focuses on the synthetic study of natural products that incorporate benzene‐fuzed chromanone (especially xanthones, (1) and chromene (2) units in their structures. Among them, partially hydrogenated xanthone blennolide C (3) was isolated from the culture broth of Blennoria sp., an endophytic fungus associated with Carpobrotus edulis, along with dimeric xanthone secalonic acid B (4) [3]. Teretifolione B (5) and its methyl derivative 6, monomeric benzochromenes, were isolated from the Australian plant Conospermum teretifolium, along with conocurvone (7), a trimeric compound with anti‐HIV properties [4, 5]. In this account, we present our efforts toward the total synthesis of monomeric benzochromanones, including xanthones, its related chromanone lactones (CLs), and benzochromenes [6, 7, 8].
Representative benzochromanone and benzochromene natural products.
Synthetic Study of Xanthone Blennolide C and Chromanone Lactone Gonytolide C
2
Xanthones (1) (a type of benzochromanone) are a class of natural substances found in plants, actinomycetes, fungi, and lichens. A particular subset, characterized by the partial hydrogenation or oxygenation of one of the benzene rings, including those with dimeric structures, has attracted attention because of its unique structural characteristics and biological activities. Xanthone dimers, referred to secalonic acid B (4) [9], are metabolites of Claviceps purpurea and the series of the secalonic acids are noted for their biological properties, such as anticancer [10] and antimicrobial activity [11]. The monomeric units of these dimers were identified as the blennolide class by Zhang et al. [3] Additionally, chromanone lactones (CLs) have been reported to be related to these xanthones. For instance, Kikuchi et al*.* [12] reported the isolation of gonytolide C (9) and the corresponding dimer gonytolide A (10) (Figure 2) from the fungus Gonytrichum sp., the latter of which exhibited innate immunosuppressive properties. The biosynthetic pathway of blennolide C (3) and gonytolide C (9) has been proposed as part of neosartorin biosynthesis (Scheme 1) [13, 14]. Chrysophanol (11), originating from emodin (12), undergoes Baeyer‐Villiger oxidation and ring‐opening, followed by esterification to form moniliphenone (13), which is converted to blennolide C (3) through aromatic ring epoxidation, xanthone ring formation, and reduction. Moreover, gonytolide C (9) is thought to be derived from keto‐form 3′ of blennolide C (3) to 1,4‐epoxide 14, and then, through a retro‐Dieckmann reaction [14, 15], transformed into gonytolide C (9).
Structure of gonytolides C (9) and A (10).
Proposed biosynthesis of blennolide C (3) and gonytolide C (9).
Previously, the total synthesis of related xanthones and CLs has been reported [15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31]. Among them, asymmetric total synthesis of gonytolide C (9) was reported [23, 24], however total synthesis of blennolide C (3) was achieved only the racemic forms [23]. For example, Sudhakar et al*.* reported the total synthesis of racemic blennolide C (3) and optically active gonytolide C (9) through the Aldol reaction between acetophenone derivatives and butyrolactone containing α‐keto ester followed by diastereoselective intramolecular cyclization as key steps [23]. Our strategy was designed not only for the total synthesis of blennolides but also for their derivatives with varying ring sizes. The construction of asymmetric quaternary stereocenters is a significant challenge in the synthesis of natural xanthone derivatives. Spiro compounds, a class of natural products and synthetic intermediates, feature quaternary carbon centers [32, 33]. Blennolide C (3) and gonytolide C (9) with an asymmetric quaternary carbon center can be synthesized via spirochromanone intermediates 15. The synthesis of the latter involves the Aldol reaction of modified acetophenones 16 with α‐oxygenated cyclohexenone 17 to form adducts 18, followed by cyclization. The xanthone framework is constructed through the oxidative cleavage of the alkene moiety in spirochromanone 15 and Dieckmann condensation of the chromanone and ester components in the side chain of chromanone ester 19. The use of cycloalkenones other than 6‐membered rings yields derivatives with different ring sizes. Lactone formation from alcohol 20 (R = OH) can lead to CL gonytolide C (9) (Scheme 2).
Retrosynthetic analysis of blennolide C (3) and gonytolide C (9).
Synthetic Studies Toward Blennolide C and Gonytolide C
2.1
We commenced our synthetic study of (±)‐4‐deoxy derivative 21 of blennolide C (3), using simple cyclohexenone (22). Friedel–Crafts acylation of 5‐methylresorcinol (23) with acetyl chloride using AlCl_3_ followed by mono protection as MOM ether gave hydroxyacetophenone 24. The Aldol reaction of 24 and cyclohexenone (22) using two equivalents of LDA afforded Aldol adduct 25, which was treated with HCl in methanol to yield spirochromanone 26. Subsequent Lemieux‐Johnson oxidation of the cyclohexene moiety and Jones oxidation of the corresponding dial, followed by ester formation yielded diester 27. This compound was treated with TiCl_4_ in the presence of Et_3_N [19] to afford the desired racemic or (±)‐4‐deoxyblennolide ((±)‐21) along with its corresponding methyl enol ether (Scheme 3) [34].
Total synthesis of (±)‐4‐deoxyblennolide C ((±)‐21).
Next, we examined the synthesis using optically active α‐oxygenated cyclohexenone 17 and simple o‐hydroxyacetophenone (28). Racemic acetate (±)‐29 was subjected to enzymatic kinetic resolution [35] to afford optically active acetate (S)‐29 with the desired absolute configuration and high optical purity (>99%ee). However, trials for the hydrolysis of acetate 29 were not successful under the acidic and basic conditions we examined; therefore, alcohol (R)‐30 with a lower ee (36% ee) generated by enzymatic resolution was used for further experiments. The application of benzyloxycyclohexenone (R)‐17 derived from alcohol (R)‐30 was subjected to an Aldol reaction with acetophenone 28, affording Aldol adduct 31 as a mixture of diastereoisomers. This was further treated with HCl in methanol to afford spirochromanones 32 and 33 as a mixture of diastereomers. The separated spirochromanone 33 with the desired relative configuration was subjected to oxidative cleavage of the alkene part followed by esterification to afford diester 34. Treatment of 34 with the TiCl_4_/Et_3_N system afforded deprotected alcohol 35, which was treated with p‐TsOH to afford CL 36, the model CL of gonytolide C (9). Dieckmann cyclization of 35 using TiCl_4_ in refluxed 1,2‐dichloroethane afforded demethylated blennolide C (37) (Scheme 4).
Total synthesis of demethylated blennolide C (37) and gonytolide C (36).
To achieve the total synthesis of natural products, synthetic procedure of optically active cyclohexenone 17 was redesigned, using the enzymatic resolution of racemic benzyloxycyclohexanol (±)‐38 [36], followed by Swern oxidation and Larock‐type dehydrogenation [37]. A similar procedure for the construction of diester 19 was adopted using acetophenone 5 and cyclohexenone 17 with (S) configuration. Treatment of diester 19 with TiCl_4_/Et_3_N afforded (+)‐blennolide C (3), its methyl enol ether 40, and deprotected alcohol 20. The latter 20 was then converted to gonytolide C (9) using p‐TsOH in refluxed toluene (Scheme 5). As a result, the first total synthesis of optically active blennolide C (3) was achieved [38].
Total synthesis of blennolide C (3) and gonytolide C (9) via spirochromanone 15.
Synthetic Study for Benzochromene Teretifoliones
3
Benzochromenes (pyranonaphthalenes) have been isolated from various plants (Figure 3) [39]. Among them, busseihydroquinones B‐D (41–43) and pyranonaphthalene 44, isolated from Pentas bussei, were reported as antiplasmodial compounds [40]. Pentacyclic busseihydroquinone E (45), the corresponding ethoxy derivative parvinaphthol C (46), and dimeric benzochromene 47 were isolated from P. parvifolia [40]. Several monomeric benzochromenes, including teretifolione B (5) and its methyl derivative 6 were isolated from Conospermum teretifolium [40]. Further research on the activity‐guided isolation of the same plant resulted in the isolation of a trimeric analog, conocurvone (7). Notably, the trimeric conocurvone (7) demonstrated anti‐HIV‐1 activity; however, the monomeric teretifolione B shows no such activity [4, 5]. Several groups have been involved in the synthesis of conocurvone (7) and related monomeric species. The semisynthesis of conocurvone (7) from isolated teretifolione B (5) was reported by Boyd et al*.* [4, 5] to elucidate the structural correlation. The enantioselective total synthesis of teretifolione B (5) was reported by Vander Velde and Jacobsen through the kinetic resolution of racemic chromenes [41]. Methylteretifolione B (6) was synthesized in racemic form by Stagliano et al., via regioselective directed ortho metalation for the introduction of methylgroup [42].
Representative benzochromene natural products 41–47.
Our synthetic study of teretifolione B (5) is based on the preparation of optically active benzochromene 48 and the Diels–Alder reaction (DAR) of pyranobenzyne 49 [43, 44, 45] and oxygenated furans 50, which are converted from tetronic acids 51 (Scheme 6a). In the future, trimeric furan 52 (Scheme 6b) could be adopted in DAR with three equivalents of benzyne 49 for the synthesis of trimeric benzochromene conocurvone (7). Here, we provide an overview of the synthetic studies on teretifoliones 5 and 6. We first explored DAR using methylated furan 50 (R = CH_3_) for the synthesis of methylteretifolione B (6) because the asymmetric total synthesis of 6 had not been reported at that time.
(a) Retrosynthesis of teretifoliones B (5, 6). (b) Structure of trimeric furan as a presumed diene for DAR.
Synthesis of Model Naphthoquinones via Diels–Alder Reactions of Benzynes and Furans
3.1
We examined the synthesis of siloxyfuran 53 (R = CH_3_) and DAR using simple benzyne. O‐Selective benzylation of methyltetronic acid (54) was achieved using BnOTs, and the resulting tetronate was converted to siloxyfuran 53. Treatment of siloxyfuran 53 with benzyne generated from triflate 55 and n‐BuLi afforded benzylated hydroxynaphthoquinone, which was treated with FeCl_3_ to convert to naphthoquinone 56 and naphthol pthiocol (57) (Scheme 7) [46].
Synthesis of naphthoquinones 56 and 57 via DAR of benzyne and furan 53.
Chromene‐derived benzyne precursor 58 with 2,2‐dimethyl groups was prepared as follows: commercially available resorcinol monobenzoate (59) was converted to iodinated propargyl ether 60. The Claisen rearrangement of 60 afforded chromene 61, and the subsequent ester exchange yielded iodo triflate 58 as a benzyne precursor. Treatment of 58 with n‐BuLi in the presence of furan 53 afforded two regioisomeric DA adducts 62 and 63, in which the desired regioisomer 62 was obtained as a minor component. Both adducts were treated with FeCl_3_ or PIDA, followed by debenzylation using BCl_3_ to afford chromenoquinone 64 with the desired regiochemistry and its regioisomer 65 (Scheme 8) [47].
Synthesis of naphthoquinones 64 and 65 via DAR of benzyne precursor 58 and furan 53.
Total Synthesis of Teretifoliones
3.2
Next, we examined the synthesis of optically active methylteretifolione B (6). Optically active chromene acetate 66 with a 4‐methypent‐3‐enyl substituent was prepared by the condensation of citral (67) and resorcinol (68) in the presence of ethylenediamine using a modified reported procedure [48], followed by the enzymatic resolution of racemic acetate (±)‐66 using Amano PS in the presence of t‐BuOH [49]. The application of hexanoate or chloroacetate instead of acetate in 66 afforded the ester with a lower ee [50]. After the conversion of the acetate to iodo triflate 48 in a similar manner, n‐BuLi‐mediated DAR with siloxyfuran 53 was performed, affording regioisomeric Diels–Alder adducts 67 and 68 in 26% and 45% yields, respectively. Both regioisomers were subjected to hydroquinone oxidation and benzyl ester hydrolysis to afford methylteretifolione B (6) and its regioisomer 69. This result was the first example of total synthesis of optically active methylteretifolione B (6) (Scheme 9). In the course using siloxyfuran 70 without a methyl group, the total synthesis of teretifolione B (5) was also achieved [51].
Synthesis of methylteretifolione B (6) and its regioisomer 69 via DAR of benzyne precursor 48 and furan 53.
Solvent and Temperature Effects on the Regioselectivity of DAR
3.3
The effects of the solvent and temperature on the regioselectivity of DAR were examined using racemic iodo triflate (±)‐48 and siloxyfuran 70 (Table 1). The reaction under the original conditions (in THF, −78°C) afforded desired benzochromene 71 and undesired 72 in a 1:1.7 ratio, in which the undesired isomer was preferential (run 1). The regioselectivity was independent of temperature (runs 1–3). In the reaction in various solvents, DME gave a result similar to that of THF (run 4); however, the reactions in less polar solvents, such as hexane and toluene, resulted in an increase in the ratio of undesired 72 (runs 5 and 6). We speculate that the regioselectivity is primarily governed by the electrostatic interactions between the electron‐rich 5‐position of furan 70 and the electrophilic 6‐position of benzyne 49, favoring the formation of undesired isomer 72. Although steric repulsion exists between the side chain of benzyne and the bulky TIPS group in furan, it is overridden by the electrostatic effect (TS1 > TS2) (Figure 4a,b). In less polar solvents, a conformational change in the benzyne side chain from “closed” to “open” conformation reduces steric hindrance between furan 70 and the side chain in 49, thus leading to further increase of the formation of 72 (TS3 > TS1) (Figure 4a,c) [51].
Plausible transition state models on DAR with benzyne 49 and furan 70. (a) The model to afford undesired cycloadduct 72 with benzyne 49 in the “closed” confirmation. (b) The model to afford desired cycloadduct 71 with 49 in the “closed” confirmation. (c) The model to afford undesired cycloadduct 72 with benzyne 49 in the “open” confirmation.
Summary and Outlook
4
In summary, this research has significantly advanced the total synthesis of benzochromanone and benzochromene natural products, exemplified by the successful construction of complex frameworks such as blennolides, gonytolide, and teretifoliones. The development of a versatile synthetic strategy utilizing spirochromanone intermediates, the achievement of total syntheses of key natural products, and the optimization of Diels–Alder and enantioselective processes have collectively broadened access to a diverse array of bioactive compounds. These accomplishments not only demonstrate the power and flexibility of the developed methodologies but also lay a strong foundation for future exploration.
Conflicts of Interest
The author declares no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A. Gaspar , E. M. P. J. Garrido , F. Borges , and J. M. P. J. Garrido , “Biological and Medicinal Properties of Natural Chromones and Chromanones,” ACS Omega 9 (2024): 21706.38799321 10.1021/acsomega.4c 00771 PMC 11112580 · doi ↗ · pubmed ↗
- 2Y. Xi , H. Wang , L. Sun , X. Ma , S. Zhang , and Z. Zhang , “Recent advances in the structures and bioactivities of benzopyrans derived from marine fungi: a review,” Frontiers in Pharmacology 15 (2024): 1482316.39512833 10.3389/fphar.2024.1482316 PMC 11540774 · doi ↗ · pubmed ↗
- 3W. Zhang , K. Krohn , U. Flörke , et al., “New Mono‐ and Dimeric Members of the Secalonic Acid Family: Blennolides A–G Isolated from the Fungus Blennoria sp.,” Chemistry–A European Journal 14 (2008): 4913.18425741 10.1002/chem.200800035 · doi ↗ · pubmed ↗
- 4L. A. Decosterd , I. C. Parsons , K. R. Gustafson , et al., “HIV inhibitory natural products. 11. Structure, absolute stereochemistry, and synthesis of conocurvone, a potent, novel HIV‐inhibitory naphthoquinone trimer from a Conospermum sp.,” Journal of the American Chemical Society 115 (1993): 6673.
- 5J. R. Dai , L. A. Decosterd , K. R. Gustafson , J. H. Cardellina , G. N. Gray , and M. R. Boyd , “Novel Naphthoquinones from Conospermum incurvum ,” Journal of Natural Products 57 (1994): 1511.7853001 10.1021/np 50113 a 006 · doi ↗ · pubmed ↗
- 6K. Katakawa and T. Kumamoto , “Chemistry of Anti‐HIV Active Trimeric Benzochromene Conocurvone: Synthetic Studies towards Monomeric Teretifolione B and Related Compounds,” Journal of Synthetic Organic Chemistry Japan 76 (2018): 722
- 7K. Katakawa and T. Kumamoto , “Chemistry of Anti‐Hiv Active Trimeric Pyranonaphthoquinone Conocurvone: Synthetic Studies towards Monomeric Teretifolione B and Related Compounds,” Heterocycles 100 (2020): 177.
- 8B. Franck , E. M. Gottschalk , U. Ohnsorge , and G. Baumann , “The Structure of Secalonic Acids A and B,” Angewandte Chemie, International Edition in English 3 (1964): 441.