A Case of IDH‐Mutant Astrocytoma Harboring an IDH2 R172_H173delinsSN Variant
Thomas Auen, Jie Chen, Nicole Shonka, Sahara Cathcart

TL;DR
A 32-year-old man with a brain tumor had an unusual IDH2 mutation, leading to a grade 3 astrocytoma diagnosis with no tumor progression after treatment.
Contribution
Presentation of a previously unreported IDH2 variant in an IDH-mutant astrocytoma case.
Findings
The tumor had a novel IDH2 (p.R172_H173delinsSN) variant and retained ATRX expression.
Methylation profiling confirmed the tumor as an IDH glioma, subclass astrocytoma.
The patient showed no tumor progression 32 months after treatment.
Abstract
IDH‐mutant gliomas most commonly harbor the canonical IDH1 p.R132H mutation, followed by less common mutations involving IDH1 p.R132 or IDH2 p.R172 codons. We present a case of a 32‐year‐old male found to have a left temporal brain tumor with regional enhancement on brain MRI, for which he underwent resection. Histologic sections showed an infiltrating astrocytic tumor with increased mitotic activity and elevated Ki67 (MIB1) labeling. The tumor was negative for IDH1 p.R132H mutant protein expression with retained ATRX expression. 1p/19q was intact by FISH analysis, and next‐generation sequencing identified a previously unreported IDH2 (p.R172_H173delinsSN) likely pathogenic variant. A diagnosis of astrocytoma, IDH‐mutant, CNS WHO grade 3 was rendered. Subsequent tumor methylation profiling performed at the National Institutes of Health matched with high confidence to the class “IDH…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
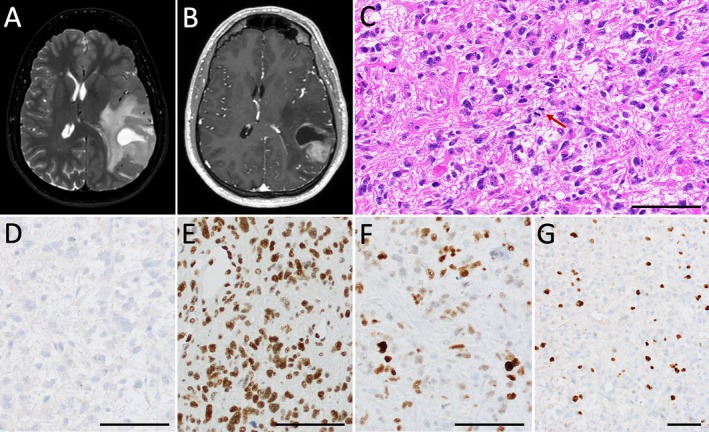 FIGURE 1
FIGURE 1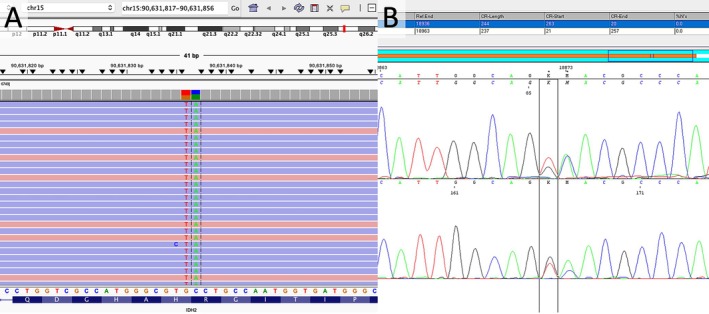 FIGURE 2
FIGURE 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlioma Diagnosis and Treatment · Meningioma and schwannoma management · Brain Metastases and Treatment
Introduction
1
IDH‐mutant astrocytoma is an IDH1‐ or IDH2‐mutant adult‐type infiltrating glioma with the absence of 1p/19q codeletion, frequent ATRX (alpha‐thalassemia/mental retardation, X‐linked) and TP53 (tumor protein p53) mutations, and frequent MGMT (O6‐methylguanine‐DNA methyltransferase) promoter hypermethylation. The vast majority, reportedly 89%–93%, of IDH‐mutant gliomas harbor the canonical IDH1 p.R132H mutation, followed by IDH1 p.R132C, p.R132G, p.R132S, p.R132L, and p.R132K. IDH2 mutations are relatively uncommon, accounting for approximately 3% of all IDH1 and IDH2 mutations in glioma, occurring more frequently in oligodendrogliomas, with p.R172K being the most frequent, and rare cases harboring p.R172L, p.R172M, p.R172W, p.R172S, p.R172G, and p.R172I [1, 2], and further exceptionally rare reports of gliomas harboring IDH2 p.P158L and p.P162S [3]. We present a case of astrocytoma, IDH‐mutant, CNS WHO grade 3 with a previously unreported IDH2 (p.R172_H173delinsSN) variant confirmed by sequencing studies and supported by DNA methylation profiling.
Case Presentation
2
The patient is a 32‐year‐old male who presented with headaches, nausea, and vomiting and was found to have a 4.2‐cm, T2/FLAIR hyperintense, solid and cystic left posterior temporal mass with regional enhancement on magnetic resonance imaging (MRI) (Figure 1A,B). He underwent left frontotemporal parietal craniotomy for resection with awake cortical language mapping. Neuropathological examination demonstrated an infiltrating astrocytoma with nuclear irregularity and hyperchromasia (Figure 1C–G). A prominent delicate vascular network was present without definitive microvascular proliferation. No necrosis was identified. Foci of perivascular inflammation were present. At least 8 mitoses per 10 high power fields (3.5 per mm^2^) were identified, and the Ki67 proliferation index was elevated at approximately 10%–15%. By immunohistochemistry, the tumor was negative for IDH1 p.R132H mutant protein expression with retained ATRX expression. Approximately 70% of tumor cells had weak to moderate nuclear p53 overexpression. Fluorescence in situ hybridization (FISH) analysis demonstrated intact chromosome 1p36 and 19q13 loci and absence of CDKN2A (cyclin‐dependent kinase inhibitor 2A) (9p21) homozygous deletion. Gain of the 9p21 locus was seen in most tumor cells. Next‐generation sequencing (NGS; Precision Oncology Profile 300, adapted from Illumina TruSight Oncology 500 panel, https://www.testmenu.com/nebraska/Tests/1224493) was performed with an estimated tumor purity of approximately 80%. Likely pathogenic mutations were identified in TP53 (p.H179R and c.994‐2A > G) at variant allele frequencies (VAF) of 45% and 46%, respectively, as well as a previously uncharacterized IDH2 (c.516_517delGCinsTA; p.R172_H173delinsSN) variant at a VAF of 56% (Figure 2A). MYCL copy number gain was also identified. Notably, no variants were identified in the EGFR, PTEN, TERT, H3F3A, or ATRX genes, nor were other variants in IDH1 or IDH2 identified. MGMT testing was negative for promoter hypermethylation by pyrosequencing. The identified IDH2 variant was confirmed by dideoxy (Sanger) sequencing (Figure 2B). To further validate the novel IDH2 variant, tissue was sent to the NCI/NIH for DNA tumor methylation profiling. The tumor demonstrated a consensus match to the class “IDH glioma, subclass astrocytoma” on the NCI/Bethesda classifier and versions 11b6 and 12b6 of the Heidelberg classifier with high‐confidence scores. Dimensionality reduction with UMAP (Uniform Manifold Approximation and Projection) also placed the tumor in the same class. Additionally, absence of MGMT promoter hypermethylation was corroborated. The patient received adjuvant concurrent radiation and temozolomide. Imaging 4 months post resection was concerning for mild progression. He received six additional cycles of temozolomide over the following 6 months, after which MRI demonstrated partial response. MRI at approximately 32 months post‐resection demonstrated no evidence of interval tumor progression.
Preoperative brain MRI demonstrated a T2/FLAIR‐hyperintense solid and cystic, mass (A, T2) in the left posterior temporoparietal lobe with nodular contrast enhancement (B, T1 postcontrast). Microscopic examination demonstrated an infiltrating astrocytoma with hyperchromasia, irregular nuclear borders, and increased mitotic activity (C, H&E, 200×, mitosis highlighted by red arrow) without definitive vascular proliferation or necrosis identified. Tumor cells were negative for IDH1 p.R132H mutant protein expression (D) with retained nuclear ATRX expression (E) and overexpression of p53 (F). The Ki67 (MIB1) proliferative index was elevated at approximately 10%–15% (G). Black bars (C–G) = 50 μm.
Next‐generation sequencing (Precision Oncology Profile 300, adapted from Illumina TruSight Oncology 500 panel, https://www.testmenu.com/nebraska/Tests/1224493) identified a novel IDH2 (c.516_517delGCinsTA; p.R172_H173delinsSN) variant at a VAF of 56% (A), which was confirmed by dideoxy sequencing (B).
Discussion
3
We present an IDH‐mutant astrocytoma with a somewhat atypical molecular phenotype including a previously uncharacterized IDH2 (c.516_517delGCinsTA, p.R172_H173delinsSN; NM_002168.2) variant involving a compound substitution of serine (S) and asparagine (N) in place of arginine (R) and histidine (H) at codons 172 and 173, respectively, notably involving the hotspot R172 codon. Additionally, the tumor exhibited retained ATRX expression and lack of MGMT promoter methylation, findings each documented in a minority of IDH‐mutant astrocytoma. A literature search of English‐language articles published on PubMed dating through May 2025 (keywords: “IDH” OR “IDH1” OR “IDH2” OR “isocitrate dehydrogenase” AND “glioma” AND “non‐canonical”) yielded no mention of an R172 compound substitution variant among identified non‐canonical IDH2 mutations in astrocytoma. This novel IDH2 variant has also not been reported in the ClinVar NCBI database. This described IDH2 variant has not been functionally validated, and thus its pathogenicity is not unequivocally determined. Though not definitive evidence, tumor methylation profiling with high confidence match to the class “IDH glioma, subclass astrocytoma” offers supportive data that this novel IDH2 variant is likely tumorigenic and may result in similar biochemical and epigenetic changes as seen in typical IDH‐mutant gliomas. Notably, the IDH2 R172 wildtype codon is highly conserved across vertebrate species and eukaryotic IDH2 homologs [4, 5], such that any alterations at this codon would be expected to have significance. In addition, this variant has not been identified in gnomAD browser (gnomad.broadinstitute.org), indicating no known population frequency.
While identification of an IDH1 or IDH2 mutation in the setting of glioma is critical for diagnostic accuracy, it has more recently been assessed as a therapeutic marker in gliomas. The IDH1 (cytosolic) and IDH2 (mitochondrial) isoforms of isocitrate dehydrogenase (IDH) convert isocitrate to α‐ketoglutarate to produce NADPH. Mutant forms of IDH further reduce α‐ketoglutarate to the oncometabolite D‐2‐hydroxyglutarate (D‐2‐HG), which results in downstream global DNA hypermethylation patterns, impaired immunity, and gliomagenesis [6]. Vorasidenib, a brain‐penetrant next‐generation dual inhibitor of mutant IDH1 and IDH2, validated using IDH1 p.R132H and IDH2 p.R140Q models, was demonstrated to bind analogous allosteric pockets of each mutant protein, respectively, with good potency against both isoforms [7]. It has recently been studied in an international phase 3 trial in grade 2 IDH1‐ and IDH2‐mutant gliomas (INDIGO trial, NCT04164901) and demonstrated improved median progression‐free survival (27.7 versus 11.1 months) and delayed time to subsequent intervention. Importantly, the vast majority of enrolled test patients harbored the canonical IDH1 p.R132H mutation, with only small numbers of patients harboring mutations in IDH1 p.R132C, p.R132G, p.R132L, and p.R132S, as well as only a few patients with IDH2 p.R172K and p.R172G [8]. With potential expanded future use of therapeutic IDH inhibitors to CNS tumors, recognition and future study of rare IDH1/2 mutations will be clinically important for these select patients to further establish efficacy. Though not a substitution for formal functional validation, this case also highlights how the use of DNA methylation profiling may help classify uncharacterized variants within tumor class‐defining genes.
Funding
The authors have nothing to report.
Disclosure
The authors have nothing to report.
Consent
The patient has given consent for the publication of this case.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1D. J. Brat , K. Adalpe , J. A. Bridge , et al., “Molecular Biomarker Testing for the Diagnosis of Diffuse Gliomas: Guidelines From the College of American Pathologists in Collaboration With the American Association of Neuropathologists, Association of Molecular Pathologists, and Society for Neuro‐Oncology,” Archives of Pathology & Laboratory Medicine 146, no. 5 (2022): 547–574, 10.5858/arpa.2021-0295-CP.35175291 PMC 9311267 · doi ↗ · pubmed ↗
- 2L. Poetsch , C. Bronnimann , H. Loiseau , et al., “Characteristics of IDH‐Mutant Gliomas With Non‐Canonical IDH Mutation,” Journal of Neuro‐Oncology 151, no. 2 (2021): 279–286, 10.1007/s 11060-020-03662-x.33205355 · doi ↗ · pubmed ↗
- 3J. Koh , H. Cho , H. Kim , et al., “ IDH 2 Mutation in Gliomas Including Novel Mutation,” Neuropathology 35 (2015): 236–244, 10.1111/neup.12187.25495392 · doi ↗ · pubmed ↗
- 4A. Nekrutenko , D. M. Hillis , J. C. Patton , R. D. Bradley , and R. J. Baker , “Cytosolic Isocitrate Dehydrogenase in Humans, Mice, and Voles and Phylogenetic Analysis of the Enzyme Family,” Molecular Biology and Evolution 15, no. 12 (1998): 1674–1684, 10.1093/oxfordjournals.molbev.a 025894.9866202 · doi ↗ · pubmed ↗
- 5N. K. Kloosterhof , L. B. C. Bralten , H. J. Dubbink , P. J. French , and M. J. van den Bent , “Isocitrate Dehydrogenase‐1 Mutations: A Fundamentally New Understanding of Diffuse Glioma?,” Lancet Oncology 12, no. 1 (2011): 89–91, 10.1016/S 1470-2045(10)70053-X.20615753 · doi ↗ · pubmed ↗
- 6R. Rudà , C. Horbinski , M. van den Bent , M. Preusser , and R. Soffietti , “IDH Inhibition in Gliomas: From Preclinical Models to Clinical Trials,” Nature Reviews Neurology 20, no. 7 (2024): 395–407, 10.1038/s 41582-024-00967-7.38760442 · doi ↗ · pubmed ↗
- 7Z. Konteatis , E. Artin , B. Nicolay , et al., “Vorasidenib (AG‐881): A First‐In‐Class, Brain‐Penetrant Dual Inhibitor of Mutant IDH 1 and 2 for Treatment of Glioma,” ACS Medicinal Chemistry Letters 11, no. 2 (2020): 101–107, 10.1021/acsmedchemlett.9b 00509.32071674 PMC 7025383 · doi ↗ · pubmed ↗
- 8I. K. Mellinghoff , M. J. van den Bent , D. T. Blumenthal , et al., “Vorasidenib in IDH 1‐ or IDH 2‐Mutant Low‐Grade Glioma,” New England Journal of Medicine 389, no. 7 (2023): 589–601, 10.1056/NEJ Moa 2304194.37272516 PMC 11445763 · doi ↗ · pubmed ↗