Multilocus sequence typing and genomic characterization of the novel Streptomyces strain SCPE-10, that resists 2,4-D toxicity and has bioremediation potential
Nildo Alfredo Nhampossa, João Victor Castro de Almeida Araújo, Vicente Almeida Serafim da Silva, João Ricardo Vidal Amaral, Cinara Souza da Conceição, Carlos Alberto Xavier Gonçalves, Eamim Daidrê Squizani, Sheila da Silva, Marcia Soares Vidal, Yinglong Chen, Andrew Macrae

TL;DR
A new Streptomyces strain, SCPE-10, was found to break down the herbicide 2,4-D and has potential for environmental cleanup and producing useful compounds.
Contribution
The discovery and genomic characterization of a novel Streptomyces strain with 2,4-D resistance and bioremediation potential.
Findings
SCPE-10 can use 2,4-D as its sole carbon source and degrade aromatic herbicides.
The genome contains genes for aromatic compound degradation and 28 biosynthetic gene clusters.
The strain is closely related to Streptomyces phaeoluteichromatogenes based on multilocus sequence analysis.
Abstract
The widespread use of aromatic herbicides such as 2,4-dichlorophenoxyacetic acid (2,4-D) has led to persistent environmental contamination, requiring efficient and sustainable biodegradation strategies. In this study, we isolated and characterized a novel actinobacterial strain, Streptomyces sp. SCPE-10, from contaminated coastal soil, capable of using 2,4-D as its sole carbon source. Phenotypic assays revealed robust growth on aromatic substrates, while whole-genome sequencing (Submission no. SUB15461668) followed by multilocus sequence analysis (16 S rRNA, recA, rpoB, atpD, gyrB, and trpB) revealed that SCPE-10 is closely related to Streptomyces phaeoluteichromatogenes. Functional genome annotation revealed a high abundance of genes involved in aromatic compound degradation, including cytochrome P450 monooxygenases, ring-cleaving dioxygenases, and dehalogenases. KEGG and antiSMASH…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1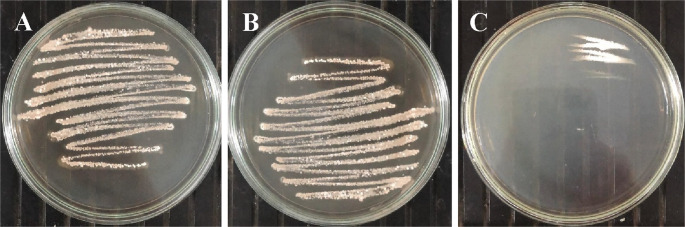 Figure 2
Figure 2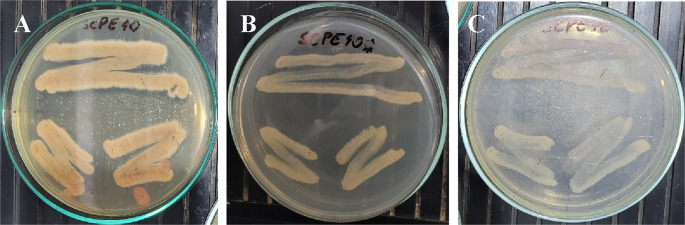 Figure 3
Figure 3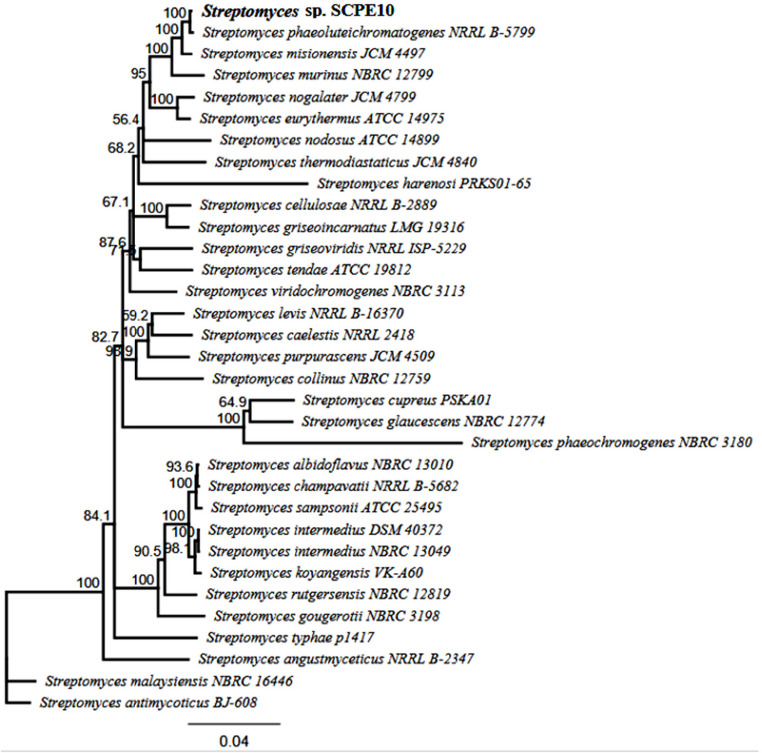 Figure 4
Figure 4- —Universidade Federal Do Rio De Janeiro
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial bioremediation and biosurfactants · Pesticide and Herbicide Environmental Studies · Microbial Natural Products and Biosynthesis
Introduction
The widespread contamination of ecosystems by aromatic compounds including, synthetic herbicides, industrial dyes, and polycyclic aromatic hydrocarbons (PAHs), represents a persistent environmental concern with direct implications for human and ecological health [1–3]. Biodegradation has emerged as a sustainable alternative for mitigating these pollutants, with members of the phylum Actinomycetota, particularly those from the genus Streptomyces, playing a central role due to their rich metabolic repertoire and ecological adaptability [4–6]. The elucidation of the molecular mechanisms underlying these catabolic abilities requires comprehensive genomic and functional analyses. Advanced bioinformatic tools such as antiSMASH have enabled the identification of biosynthetic gene clusters (BGCs), which often encode key enzymes involved in biodegradation pathways [7]. Additionally, databases like KEGG provide detailed insights into metabolic routes associated with the oxidation and cleavage of aromatic rings, involving enzymes such as monooxygenases, dehydrogenases, and dioxygenases [8–10]. In this study, we report on the complete genome sequencing and genome annotation of Streptomyces sp. strain SCPE-10 and perform a comprehensive genome mining analysis aimed at identifying genes and BGCs related to the degradation of aromatic compounds. Using a combined approach based on antiSMASH and KEGG pathway analysis, we explore the metabolic potential of SCPE-10, with particular focus on its capacity to degrade environmental pollutants of aromatic nature. This work highlights the ecological and biotechnological potential of SCPE-10 for future applications in environmental bioremediation.
Materials and methods
Microbial strain
Streptomyces sp. SCPE-10 was selected from a screening of approximately 710 actinobacteria strains from the culture collection of the Laboratory of Microbial Process Engineering at UFRJ (LEPM-EQ/UFRJ). All isolates were grown on ISP-2 at 28 °C and pH 7.0. for 10 days, and the fastest-growing strains were tested for tolerance to 250 ppm of 2,4-dichlorophenoxyacetic acid (2,4-D) under three conditions: (1) with 1.0% malt extract; (2) with 0.5% malt extract; and (3) 2,4-D as the sole carbon source. SCPE-10 showed robust growth in some conditions, including minimal medium, and was thus selected for further biodegradation and genomic analysis.
Phenotypic characterization
Colony morphology of Streptomyces strain SCPE-10 was evaluated after growth on KB and ISP-4 (International Streptomyces Project medium), incubated at 28 ± 1 °C for up to 10 days. An overnight culture from Gause #1 medium was streaked and observed for features such as color, form, and transparency. All assays were performed in triplicate.
Ability of Streptomyces SCPE 10 to degrade aromatic compounds
The ability of Streptomyces sp. SCPE-10 to degrade the herbicide 2,4-dichlorophenoxyacetic acid (2,4-D) was initially tested on solid media containing 250 ppm 2,4-D with (1) 1.0% or (2) 0.5% malt extract, and (3) 2,4-D as sole carbon source. SCPE-10 only grew in media supplemented with malt extract, suggesting co-metabolism is required. A spore suspension was prepared from ISP-2 plates, and spores were harvested, filtered, and stored in 35% glycerol (4.97 × 10^10^ CFU/mL). Submerged fermentation was performed in 250 mL medium (250 ppm 2,4-D + 1.0% or 0.5% malt extract), inoculated with 897 µL of the spore suspension, and incubated at 28 °C, 180 rpm, for 14 days. Samples were collected at defined intervals and stored for enzymatic and degradation analyses.
Genome sequencing, library construction, and annotation
Genomic DNA was extracted using phenol–chloroform, and sequenced with PacBio HiFi. Assembly was done with Hifiasm and annotated via Prokka v1.14.6. Completeness was assessed with BUSCO (Benchmarking Universal Single-Copy Orthologs) against the Actinobacteria database. Identification was confirmed by 16 S rRNA analysis using EzBioCloud (https://kb.ezbiocloud.net/home/protocols/16s-identication) and multilocus sequence analysis (MLSA) with six housekeeping genes: 16 S rRNA (16 S ribosomal RNA), recA (recombinase A), rpoB (RNA polymerase subunit beta), atpD (ATP synthase subunit beta), gyrB (DNA gyrase subunit B), and trpB (tryptophan synthase subunit beta), aligned and phylogenetically analyzed in Geneious v2025.0.3. (https://www.geneious.com). Functional annotation was performed with antiSMASH v7.0 (https://antismash.secondarymetabolites.org/) (secondary metabolites), KEGG (https://www.genome.jp/kegg/pathway.html) (xenobiotic metabolism), and MobileOG-db (mobile elements).
In silico identification of aromatic compound degradation genes
The functional annotation of the Streptomyces sp. SCPE-10 genome was carried out to identify genes involved in the degradation of aromatic xenobiotics. Genome annotation was performed using the KEGG Orthology-Based Annotation System (https://www.genome.jp/brite/ko00001) and the RAST server. Enzymatic functions related to the degradation of chlorinated aromatic compounds were investigated, with emphasis on genes associated with the catabolism of 2,4-dichlorophenoxyacetic acid (2,4-D). The search focused on coding sequences annotated as dioxygenases (including α-ketoglutarate-dependent enzymes), monooxygenases, hydrolases, and dehalogenases. Genes homologous to members of the tfd cluster were identified based on conserved domains and sequence similarity. The presence of catechol 1,2-dioxygenase and catechol 2,3-dioxygenase genes was used to infer the potential for both ortho- and meta-cleavage of aromatic rings. Secondary metabolite biosynthetic gene clusters (BGCs) were detected using antiSMASH v7.0, and their colocalization with xenobiotic degradation-related genes was assessed to explore possible functional synergies.
Data availability
The Streptomyces sp. SCPE-10 genome has been deposited and is available in the National Center for Biotechnology Information (NCBI) under the Submission number SUB15461668.
Results
Phenotypic characterization and ability to degrade aromatic compounds
After 10 days of incubation on ISP-2 medium at 28 ± 1 °C, Streptomyces sp. SCPE-10 formed buried, dry, and firm colonies with a white to beige pigmentation, typical of actinomycetes (Fig. 1A–B). The strain’s ability to metabolize aromatic compounds was assessed using ISP-2-based media supplemented with 250 ppm 2,4-dichlorophenoxyacetic acid (2,4-D) under three nutritional conditions. Robust growth was observed in Medium 1 (1.0% malt extract) and consistent growth in Medium 2 (0.5% malt extract), whereas no growth occurred in Medium 3, which contained only 2,4-D as the sole carbon source (Figs. 2A–C). This indicates the requirement of co-metabolism for 2,4-D degradation. Colony morphology remained filamentous and branched in Mediums 1 and 2, while no sporulation or expansion was noted under carbon-limited conditions (Fig. 3A–C). Among 11 screened isolates, only SCPE-10 and C2B grew consistently on 2,4-D media, with SCPE-10 selected for further genomic investigation due to superior performance.
Fig. 1. Colony morphology of Streptomyces sp. SCPE-10 on ISP-2 agar medium viewed from different angles. (A) Top view showing colony pigmentation and structure. (B) Close-up of aerial mycelium on colony surface showing typical growth after 10 days of incubation at 28 ± 1 °C
Fig. 2. Growth of Streptomyces sp. SCPE-10 on ISP-2 agar supplemented with 2,4-D under varying nutrient conditions. (A) Medium 1: 2,4-D (250 ppm) + 1% malt extract. (B) Medium 2: 2,4-D (250 ppm) + 0.5% malt extract. (C) Medium 3: 2,4-D (250 ppm) without malt extract. Plates were incubated for up to 10 days at 28 ± 1 °C
Fig. 3. Comparative analysis of filamentous growth of Streptomyces sp. SCPE-10 on media with and without 2,4-D. (A) Mycelial density in Medium 1 indicates efficient growth. (B) Reduced mycelial development in Medium 2. (C) Repressed growth in Medium 3. Plates were incubated for up to 10 days at 28 ± 1 °C
Genome sequencing analysis
The complete genome of Streptomyces sp. SCPE-10, sequenced via PacBio HiFi, consists of a linear chromosome with 8,012,345 bp and 72.3% G + C content, assembled into a single contig without plasmids. Prokka annotation predicted 7,222 genes, including 6,982 protein-coding sequences and 145 RNAs (90 tRNAs and 18 rRNAs) (Table 1). Taxonomic analysis based on 16 S rRNA via EzBioCloud revealed 99.86% similarity to Streptomyces phaeoluteichromatogenes (93.8% query coverage) (Table 2). MLSA using six housekeeping genes (16 S rRNA, recA, rpoB, atpD, gyrB, trpB) confirmed its close affiliation with S. phaeoluteichromatogenes (Fig. 4).
BUSCO analysis indicated 99.9% genome completeness. Annotation revealed genes related to xenobiotic degradation, including cytochrome P450 monooxygenases, ring-hydroxylating dioxygenases, and multicopper oxidases, as well as carbohydrate-active enzymes like chitinases and cellulases, suggesting ecological adaptability. AntiSMASH v7.0 identified 24 biosynthetic gene clusters (BGCs), including NRPS, PKS, siderophores, lanthipeptides, and terpenes, some similar to known BGCs (e.g., actinomycin D, geosmin), others potentially novel (Table 3). KEGG pathway analysis highlighted genes linked to the degradation of chlorinated aromatics, nitroaromatics, and PAHs, reinforcing the strain’s biotechnological potential (Table 4).
Table 1. Summary of genome assembly and annotation of Streptomyces sp. SCPE-10AssemblyGenome size (bp)8,012,345Number of contigs1GC content (%)72.3Genes (total)7222CDSs (total)7113CDSs (with protein)6982tRNAs90rRNAs18tmRNAs1PseudogenesNot detectedSample IDStrain SCPE 10Assembly nameStreptomyces_SCPE 10OrganismStreptomyces SCPE 10Raw reads (fastq)287 MBAssembled genome (fasta)7.7 MBTaxonomic classificationStreptomycetales 99.9%
Table 2. Taxonomic identification of SCPE-10 was refined based on 16 S rRNA sequence analysis#SpeciesGenome groupTaxonomyIden.QueryRef.(%)cov. (%)cov. (%)1 Streptomyces phaeoluteichromatogenes -Bacteria Actinobacteria Actinomycetia99.8693.80100Streptomycetales Streptomycetaceae Streptomyces 2 Streptomyces misionensis -Bacteria Actinobacteria Actinomycetia99.7995.1100Streptomycetales Streptomycetaceae Streptomyces 3 Streptomyces malaysiense -Bacteria Actinobacteria Actinomycetia99.7295.1100Streptomycetales Streptomycetaceae Streptomyces 4 Streptomyces cupreus -Bacteria Actinobacteria Actinomycetia99.1795.1100Streptomycetales Streptomycetaceae Streptomyces 5 Streptomyces diastaticus -Bacteria Actinobacteria Actinomycetia99.0395.0100Streptomycetales Streptomycetaceae Streptomyces 6 Streptomyces intermedius Streptomyces albidoavus groupBacteria Actinobacteria Actinomycetia99.0395.0100Streptomycetales Streptomycetaceae Streptomyces 7 Streptomyces levis -Bacteria Actinobacteria Actinomycetia98.9594.1100Streptomycetales Streptomycetaceae Streptomyces 8 Streptomyces glaucescens -Bacteria Actinobacteria Actinomycetia98.8995.0100Streptomycetales Streptomycetaceae Streptomyces 9 Streptomyces collinus Streptomyces iakyrus groupBacteria Actinobacteria Actinomycetia98.8994.4100Streptomycetales Streptomycetaceae Streptomyces 10 Streptomyces venetus -Bacteria Actinobacteria Actinomycetia98.8395.1100Streptomycetales Streptomycetaceae Streptomyces
Fig. 4. Maximum-likelihood phylogenomic tree based on 33 concatenated core genes, constructed using the Geneious tool, which automates the extraction and analysis of multilocus sequence typing (MLST) data from genomic sequences. The tree includes both query strains (QSs) and type strains (TSs), which serve as references. The bar indicates sequence divergence
Table 3. Secondary metabolic gene clusters predicted in the genome of Streptomyces sp. SCPE-10RegionTypeMost similar known clusterSimilarity1terpeneherbimycin APolyketide6%2redox-cofactorlankacidin CNRP + Polyketide13%3NRPS4NRPS, NAPAAstenothricinNRP: Cyclic depsipeptide13%5T3PKSherboxidienePolyketide7%6ectoineectoineOther100%7NRPS-like8melaninmelaninOther60%9siderophoredesferrioxamin B / desferrioxamine EOther83%10butyrolactone11T2PKSspore pigmentPolyketide83%12lanthipeptide-class-iiinaphthomycin APolyketide9%13terpenejulichrome Q3-3 / julichrome Q3-5Polyketide25%14T1PKS, siderophorekinamycinPolyketide22%15thiopeptide, thioamitides, NRPS, ladderaneRP-1776Polyketide + NRP: Cyclic depsipeptide10%16other, T1PKSchlorizidine ANRP + Polyketide: Modular type I11%17NRPS-likemeilingmycinPolyketide3%18RiPP-like19terpenegeosminTerpene100%20siderophoregrincamycinPolyketide: Type II + Saccharide: Hybrid/tailoring8%21betalactonebelactosin A / belactosin COther75%22terpenehopeneTerpene92%23terpene, T1PKS, NRPSisorenierateneTerpene54%24T1PKS, NRPS-like, transAT-PKS-like, lanthipeptide-class-iisalinosporamide APolyketide23%25NRPS, terpene, lanthipeptide-class-i, lanthipeptide-class-iicadaside A / cadaside BNRP28%26NRPScyclomarin DNRP8%27RiPP-like, lanthipeptide-class-iiiinformatipeptinRiPP: Lanthipeptide57%28lanthipeptide-class-iv, PKS-like, lassopeptideGE81112NRP14%
Identification of aromatic compound degradation genes in Streptomyces sp. SCPE-10
The genome of Streptomyces sp. SCPE-10 was screened in silico to investigate its potential for degrading aromatic compounds. Annotation revealed a range of genes encoding enzymes classically involved in aromatic hydrocarbon catabolism, including cytochrome P450 monooxygenases, dioxygenases (aromatic ring-hydroxylating and ring-cleaving types), DyP-type peroxidases, and multicopper oxidases. These enzymes mediate essential steps such as hydroxylation, ring cleavage, and phenolic compound oxidation of 2,4-D (Table 4).
Additionally, genes for glutathione S-transferases and epoxide hydrolases suggest a detoxification capability via conjugation and hydrolysis of electrophilic aromatic compounds. KEGG-based pathway analysis indicated genetic potential for the degradation of benzoate, phenylacetate, naphthalene, and chlorinated aromatics. Key enzymes such as protocatechuate 3,4-dioxygenase, catechol 2,3-dioxygenase, and salicylate hydroxylase were consistently annotated. Several of these genes were located within biosynthetic gene clusters (BGCs) predicted by antiSMASH, suggesting a link between secondary metabolism and xenobiotic degradation (Table 5). The combination of these features, highlights the robust genetic capacity of SCPE 10 to metabolize a wide array of persistent aromatic pollutants.
Table 4. Main genes identified in the genome of Streptomyces sp. SCPE-10 involved in the 2,4-D degradation pathwayEnzymes encoded by the tfd genes of SCPE-10ContigKEGG codeGeneFunctionAlpha-ketoglutarate-dependent dioxygenase AlkB00909K06912 tfdA Encodes oxygenase’s and dioxygenase’s, which catalyzes the first step in 2,4-D degradation by converting it into 2,4-dichlorophenol (2,4-DCP)Alpha-ketoglutarate-dependent sulfate ester dioxygenase02595 tfdA L-lysine N6-monooxygenase01973K10676 tfdB Encodes a phenol monooxygenase, which hydroxylates 2,4-DCP into 3,5-dichlorocatechol, preparing it for ring cleavageL-lysine N6-monooxygenase02752 tfdB Cytochrome P450 monooxygenase PikC03386 tfdB Cytochrome P450 monooxygenase PikC05669 tfdB Cytochrome P450 monooxygenase PikC05753 tfdB Cytochrome P450 monooxygenase PikC06179 tfdB Cytochrome P450 monooxygenase PikC06457 tfdB putative FAD-linked oxidoreductase02874 tfdB putative FAD-linked oxidoreductase YvdP03794 tfdB 3-phenylpropionate/cinnamic acid dioxygenase ferredoxin–NAD(+) reductase component01267 tfdC Encodes a catechol 1,2-dioxygenase, which performs ortho-cleavage of the aromatic ring of 3,5-dichlorocatecholHomogentisate 1,2-dioxygenase01691 tfdC Biphenyl 2,3-dioxygenase, ferredoxin component01879 tfdC 3-carboxy-cis, cis-muconate cycloisomerase07096 tfdD Encodes a hydrolase, which acts on carboxylated ring-cleavage intermediates to facilitate further breakdownAlcohol dehydrogenase00032 tfdF Encodes a dehydrogenase, which oxidizes these intermediates, completing the mineralization of the aromatic compoundNADP-dependent alcohol dehydrogenase C 205077 tfdF Bifunctional cytochrome P450/NADPH–P450 reductase06636 tfdF Bifunctional cytochrome P450/NADPH–P450 reductase06646 tfdF putative alcohol dehydrogenase AdhA00735 tfdF Alcohol dehydrogenase02702 tfdF Alcohol dehydrogenase04748 tfdF NADP-dependent alcohol dehydrogenase C 205077 tfdF Alcohol dehydrogenase05463 tfdF Alcohol dehydrogenase06744 tfdF Alcohol dehydrogenase06941 tfdF
Table 5. Analysis of aromatic compound degradation pathways predicted in the genome of Streptomyces sp. SCPE-10Degradation PathwayKEGG Map EcsGenes Detected in SCPE 10Coverage (%)Benzoate degradation via hydroxylation501224.0%Benzoate degradation via CoA ligation441431.8%Naphthalene and anthracene degradation22627.3%Toluene and xylene degradation23521.7%Chlorobenzoate degradation13323.1%Phenol degradation9333.3%Catechol ortho- and meta-cleavage14428.6%2,4-Dichlorobenzoate degradation29310.3%Styrene degradation8337.5%Ethylbenzene degradation11436.4%Dioxin degradation12325.0%1,2-Methylnaphthalene degradation17741.2%Cyclohexanol degradation (Baeyer - Villiger)10220.0%Metabolism of xenobiotics by cytochrome P4507342.9%Fluorobenzoate degradation10110.0%
Discussion
Aromatic compounds and herbicide derivatives such as 2,4-D are among the most persistent and hazardous pollutants in agricultural soils, posing significant ecological and health risks [1, 11]. Microbial biodegradation stands out as an efficient, cost-effective, and environmentally friendly approach for remediating contaminated environments [12]. Several bacterial genera, including Pseudomonas, Bacillus, Rhodococcus, Sphingomonas, and Streptomyces, have been explored for their biodegradation capabilities [13–16]. Among them, Streptomyces spp. are notable for their genetic diversity and enzymatic arsenal that allow them to degrade a wide range of complex xenobiotics [17]. In this study, we isolated and characterized the strain Streptomyces SCPE-10, which was able to grow in the presence of 2,4-D, highlighting its resilience to toxicity and adaptation to polluted environments. Genomic analysis revealed several genes associated with hydrocarbon and aromatic compound catabolism, including cytochrome P450 monooxygenases, ring-cleaving dioxygenases, hydrolases, transferases, and dehydrogenases, enzymes already known for their role in aromatic degradation pathways [18, 19]. KEGG-based annotation identified several pathways related to the degradation of benzoate, naphthalene, toluene, ethylbenzene, fluorene, and other persistent aromatics. Genes such as tfd, pcaG, catA, xylE, and ligB were found to participate in aromatic ring cleavage via catechol and protocatechuate intermediates, leading to the production of tricarboxylic acid cycle intermediates [20–22]. AntiSmash analysis revealed 28 biosynthetic gene clusters, many associated with aromatic structures or detoxification mechanisms. These included terpenes, siderophores, ectoines, lanthipeptides, and complex NRPS/PKS clusters, underscoring the ecological versatility of SCPE 10 and its potential for biotechnological exploitation, especially in producing novel metabolites [23–25]. Notably, genes such as adhP, aldH, fadB, and paaF involved in the degradation of alkenes and halogenated aromatics were also identified, supporting SCPE 10’s capability to degrade 2,4-D and related compounds. Additionally, the presence of ubiX, associated with anaerobic aromatic degradation, suggests that the strain may maintain metabolic plasticity under variable environmental conditions [26, 27].
Overall, Streptomyces sp. SCPE-10 is a promising candidate with potential for the bioremediation of contaminated soils and may serve as a platform for the development of microbial consortia targeting xenobiotic degradation in agroecosystems. Further studies should focus on the identification of end-products, transcriptomic profiling under environmental stress, and validation of degradation efficiency in mesocosm or field-scale trials.
Conclusion
Streptomyces sp. SCPE-10 exhibited a remarkable genomic capacity to degrade aromatic compounds, highlighting its biotechnological potential for environmental remediation. Genomic analysis revealed a diverse set of catabolic genes and biosynthetic clusters linked to xenobiotic metabolism, particularly enzymes such as cytochrome P450s, aromatic ring-cleaving dioxygenases, hydrolases, and multiple NRPS/PKS clusters. These findings position SCPE-10 as a promising microbial resource for bioremediation strategies targeting herbicide-contaminated environments.