Steroid hormones and nephrolithiasis: regulation of urine components metabolism and inflammation
Xinrong Zhang, Shuaibin Wang, Jiaxin Zhao, Bingyu Xiang, Mingxia Zhang

TL;DR
This paper explores how steroid hormones influence kidney stone formation, explaining gender differences in risk and suggesting new treatment approaches.
Contribution
It systematically demonstrates that steroid hormones are key regulators of gender differences in nephrolithiasis.
Findings
Steroid hormones modulate calcium, oxalate, and phosphate metabolism related to kidney stones.
They influence the renal inflammatory microenvironment linked to stone formation.
The paper highlights potential mechanisms and target sites for steroid hormone therapy in nephrolithiasis.
Abstract
The global incidence of nephrolithiasis has increased significantly in recent decades. The prevalence remains higher in males than females, the exact mechanisms responsible for this gender-based disparity in nephrolithiasis risk remain incompletely understood. Although dietary and lifestyle factors contribute to this difference, they do not entirely account for the observed variation. Emerging evidence suggests that steroid hormones may play a pivotal role in modulating renal stone formation through their influence on calcium, oxalate, and phosphate metabolism, as well as regulating the renal inflammatory microenvironment. This review synthesizes current knowledge on the interplay between steroid hormones and nephrolithiasis pathogenesis, providing a theoretical framework for understanding gender-specific susceptibility and highlighting potential avenues for tailored preventive and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
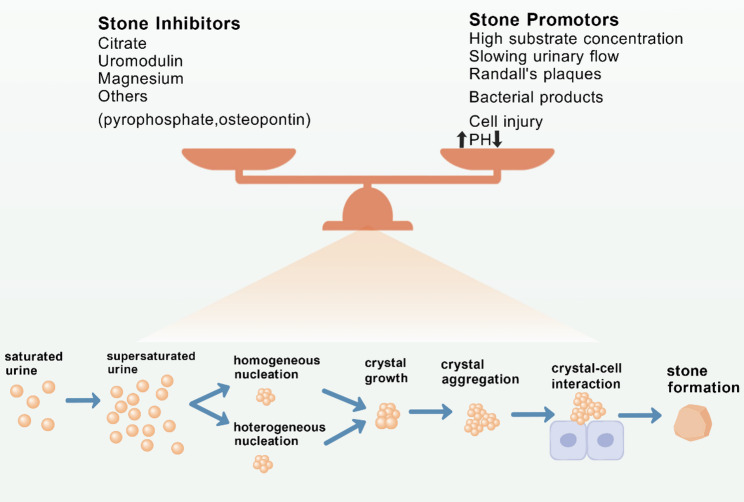 Figure 1
Figure 1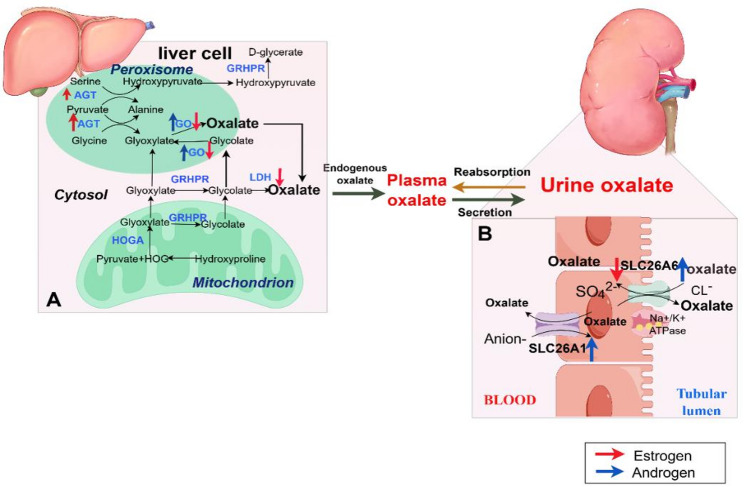 Figure 2
Figure 2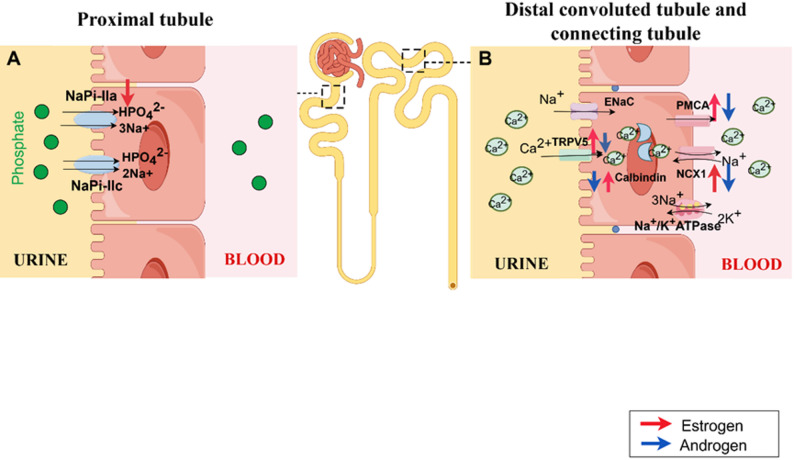 Figure 3
Figure 3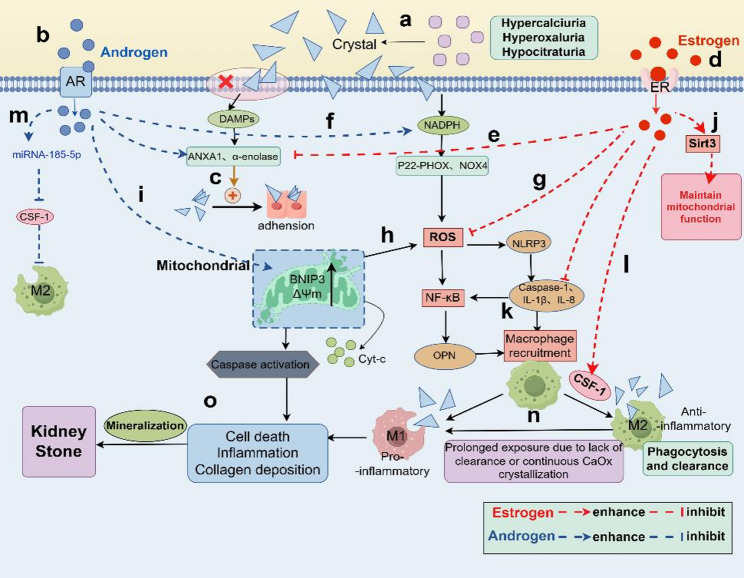 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsKidney Stones and Urolithiasis Treatments · Biomedical Research and Pathophysiology · Ureteral procedures and complications
Introduction
Nephrolithiasis is a urological disease with high global prevalence and significant recurrence rate [1]. Calcium-containing stones, primarily composed of calcium oxalate and calcium phosphate, are the most common type [2, 3]. Stone formation arises not from a single factor, but from the interplay of genetics [4], metabolism [5], environment [6], and diet [7]. Recent studies suggest that nephrolithiasis should be regarded as a systemic disease [8, 9].
Data from the U.S. National Health and Nutrition Examination Survey (NHANES) revealed a significant gender difference in nephrolithiasis incidence (6.5% in females vs. 9.3% in males) between 2008 and 2018 [10, 11]. However, other studies also suggest this gender gap in stone disease is narrowing [12, 13]. In China, a large cross-sectional study further demonstrated that the risk of nephrolithiasis increases steadily with age in females, while in males, the risk rises initially and then declines, which presumably associated with weakened androgen signaling [14]. In addition to potentially influencing social behaviors, such as dietary preferences and occupational exposure [15, 16], while steroid hormones are increasingly recognized to regulate the metabolic homeostasis of lithogenic substances and the local renal microenvironment [17, 18].
This review aims to systematically organize and deeply explore nephrolithiasis and steroid hormones of multidimensional mechanisms. By synthesizing these insights, this review seeks to advance understanding of gender differences in nephrolithiasis and inform the development of gender-specific prevention and treatment strategies.
Nephrolithiasis and metabolism of urine components: oxalate, phosphate, and calcium
The formation of kidney stones is a complex biologic process (Fig. 1) [19]. Such a process starts with urine supersaturation, followed by crystal nucleation, growth, aggregation, and retention in the kidney [20, 21]. Urinary solute supersaturation provides the necessary milieu for stone formation. So, metabolic imbalances in urine involving oxalate, phosphate, and calcium are crucially important in this context [22].
Fig. 1. Overview of the pathogenesis of nephrolithiasis
Oxalate is a core component of calcium oxalate stones, with exogenous sources (such as spinach, amaranth and other foods rich in oxalic acid) and endogenous synthesis (primarily in the liver) [23]. Under physiological circumstances, urinary oxalate concentrations are extremely low. However, metabolic disturbances can trigger a sharp rise in urinary oxalate [24]. This leads to rapid combination with calcium ions, forming highly stable calcium oxalate crystals that facilitate heterogeneous nucleation and growth into the most common type of kidney stones [8, 25].
Phosphate is essential for calcium phosphate stones and Randall’s plaque [8]. In an alkaline urinary environment, the solubility of calcium phosphate decreases, promoting precipitation [26]. Conditions like renal tubular acidosis can create such an environment persistently, fostering Randall’s plaque formation at renal papillae tips [27]. These plaques act as nucleation sites for calcium oxalate crystals, indirectly promoting stone development [8, 28].
Calcium serves as the primary substrate for calcium-containing stones. Under physiological conditions, most filtered calcium is reabsorbed, with minimal urinary excretion [29, 31]. Abnormalities in calcium metabolism, such as excessive intestinal absorption [32], impaired renal reabsorption function [33], or abnormal parathyroid hormone secretion [34], can lead to hypercalciuria. This not only provides core material for stone formation but also reduces the solubility of anions like oxalate, facilitating crystallization [35, 36].
Accordingly, this review will examine the relationship between steroid hormones and nephrolithiasis through the lens of these three key components.
Steroid hormones and nephrolithiasis: regulation of oxalate metabolism
Oxalate metabolism and nephrolithiasis
Transmembrane oxalate transport is mediated by specific transporters, with SLC26A6 and SLC26A1 (Sat-1) being key regulators of intestinal absorption and renal excretion [37]. SLC26A6, a conserved anion transporter (Cl⁻/oxalate) primarily localized to the apical membrane of intestinal epithelial cells and renal proximal tubules, is responsible for intestinal oxalate absorption and renal oxalate secretion [37]. In male SLC26A6 knockout (SLC26A6–/–) mice is demonstrated, which elevated serum and urinary oxalate levels and develop calcium oxalate urolithiasis [38]. Sat-1, a sodium-independent sulfate anion transporter (Oxalate-SO₄²⁻/HCO₃⁻) expressed in the basolateral membrane of renal proximal tubules and the sinusoidal membrane of hepatocytes, is also critical for oxalate homeostasis [39]. Previously, characterization of the Sat1–/– mouse suggested that the loss of Sat1-mediated oxalate secretion by the intestine was responsible for the hyperoxaluria, hyperoxalemia, and calcium oxalate urolithiasis reportedly displayed by this model [40]. However, recent trans-epithelial flux assays using Sat1–/– mice suggest that its regulatory role may be confined to the liver or kidneys. Additionally, Sat1–/– mice were neither hyperoxaluric nor hyperoxalemic. Instead, 24 h urinary oxalate excretion was almost 50% lower than in WT mice [41].
The liver is the primary site of endogenous oxalate synthesis, where the balance between oxalate-producing and detoxification pathways directly determines oxalate output (Fig. 2). Primary hyperoxaluria (PH), a rare genetic disorder caused by mutations or defects in liver enzymes, is the primary genetic factor leading to severe calcium oxalate kidney stones and comprises three subtypes [42, 43]. PH1 is characterized by deficiency of alanine-glyoxylate aminotransferase (AGT) [44]. In vivo studies explored the pathway of inhibiting hydroxyfumarate production by targeting hydroxyfumarate oxidase 1 (HAO1) mRNA via Dicer substrate small interfering RNA (DsiRNA), thereby reducing calcium oxalate deposition in PH1 mouse models [45]. HAO1 encodes glycolate oxidase (GO), which catalyzes the oxidation of glycolate into glyoxylate (the direct precursor of oxalic acid). PH2 deficiency involves both carboxymalonic reductase and glyoxylate reductase (GRHPR) [46]. Defective activity in either AGT or GRHPR leads to insufficient removal of glyoxylate, which is then oxidized to oxalate via lactate dehydrogenase (LDH). Increased oxalate synthesis triggers calcium oxalate deposition in kidney [47, 48]. PH3 (mitochondrial 4-hydroxy-2-ketoglutarate aldolase HOGA1 gene mutation) presented clinically with kidney stones disease at age 13 and renal failure at age 33 [49].
Fig. 2. Role of steroid hormones in oxalate metabolism (A) Endogenous pathways for oxalate synthesis in hepatocytes. Estrogen (red arrow) promotes AGT while inhibiting GO and LDH, thereby suppressing oxalate synthesis. Androgen (blue arrow) enhances GO, increasing oxalate production. (B) Oxalate transport in the renal proximal tubule. Estrogen (red arrow) reduces SLC26A6 transport, decreasing oxalate secretion, while androgen has the opposite effect. Androgen (blue arrow) enhances SLC26A1 transport, increasing blood oxalate concentration. AGT, alanine-glyoxylate aminotransferase; GO, glycolate oxidase; GRHPR, glyoxylate reductase; LDH, lactate dehydrogenase; HOGA, 4-hydroxy-2-oxoglutarate aldolase
Role of steroid hormones in oxalate metabolism
A 24-hour urine analysis of 628 kidney stone formers using gas chromatography-mass spectrometry (GC-MS) revealed a positive correlation between urinary oxalate excretion and the excretion of six androgen metabolites [50].
In vivo studies have further elucidated this regulatory relationship. In SLC26A6–/– rats, renal SLC26A6 mRNA and protein expression exhibit male-dominant patterns, and prepubertal rats show low, sex-independent expression, while adult males display high expression [51]. Castration of adult male rats reduces SLC26A6 expression, which is restored by testosterone treatment, indicating that post-pubertal androgens regulate the male-dominant expression of renal SLC26A6 [51]. Conversely, in vitro experiments indicate that estrogen (primarily β-estradiol) reduces SLC26A6 activity in the nephrons, leading to decreased transcellular oxalate secretion and reduced apoptosis rates [52]. Simultaneously, estrogen may also inhibit oxalate absorption by upregulating SLC26A6 expression or function in the intestine [53], or through other intestinal secretion/barrier mechanisms, thereby lowering blood oxalate concentrations. This explains why females typically exhibit smaller increases in urinary oxalate than males following consumption of equivalent high-oxalate diets, reducing oxalate deposition probability [50, 54]. Transport studies in isolated matrix vesicles (which are thought to initiate renal crystal deposition by inducing nucleation and crystallization) confirm that male rats have increased Sat-1 protein expression in the basolateral invaginations of cortical proximal convoluted tubules, contributing to enhanced oxalate biosynthesis [55].
Additional research has underscored the role of steroid hormones in regulating oxalate-synthesizing enzymes. In Sprague-Dawley rats [56], testosterone treatment (in both males and females) significantly increased renal crystal deposition, urinary oxalate excretion, and hepatic/renal GO expression. Conversely, estradiol treatment and male castration reduced these parameters. Another study found that Hao1 mRNA expression is predominantly male-specific and unaffected by ethylene glycol (EG) treatment [57]. Collectively, these data indicate that testosterone promotes renal crystal deposition by upregulating GO expression and enhancing oxalate synthesis, while estradiol acts as an inhibitor. Furthermore, hepatocytes serve as a major site for hepatic enzyme production. In vitro experiments further show that estradiol inhibits LDH production in hepatocytes, protecting hepatocytes against oxidative damage and apoptosis [58].
Liang et al. [59] discovered that androgen receptor (AR) signaling directly upregulates GO in the liver and the NADPH oxidase subunit P22-PHOx in renal epithelium at the transcriptional level, potentially increasing oxalate biosynthesis. In vitro studies [60] indicate that estrogen receptor β (ERβ) reduces hepatic oxalate biosynthesis by increasing AGT expression in hepatocytes, thereby inhibiting oxalate accumulation and preventing nephrolithiasis formation.
In summary, estrogen increases transporter activity and inhibits hepatic oxalate biosynthesis, whereas androgens exert the opposite effect. This can lead to differences in nephrolithiasis risk between males and females (Fig. 2).
Steroid hormones and randall’s plaques: regulation of phosphate metabolism
Phosphate metabolism and randall’s plaque formation
First identified by Alexander Randall in 1937 [61], Randall’s plaques serve as the initiation platform for calcium oxalate stones, forming through three stages: initial calcium phosphate (CaP) crystal formation, progression of interstitial biomineralization, and plaque enlargement with epithelial penetration, ultimately providing attachment sites for calcium oxalate stones development [62].
Hyperphosphatemia directly increases the saturation of calcium and phosphate ions in urine, promoting the formation of stones such as apatite and calcitrinite [63]. Phosphate transport in renal and small intestinal epithelial cells is mediated by type II Na-pi co-transporters of the SLC34 family (IIa, IIb, IIc) [64]. Intestinal phosphate absorption is primarily mediated by NaPi-IIb [65], while renal phosphate reabsorption from tubular filtrate is jointly regulated by NaPi-IIa and NaPi-IIc expressed on the brush border membrane of proximal tubule cells [66] (Fig. 3). Although NaPi-IIc contributes to phosphate reabsorption and bone mineralization, clinical data do not clearly link it to nephrolithiasis pathogenesis. Moreover, in NaPi-IIc–/– mice, phosphate homeostasis shows no significant abnormalities, and no stones are found in their kidneys [67]. In contrast, NaPi-IIa–/– mice exhibit hypophosphatemia, increased urinary phosphate excretion, and Randall’s deposits in renal tubules and interstitium [68].
Serum phosphate levels are also regulated by parathyroid hormone (PTH), Klotho (KL), and 1,25-dihydroxyvitamin D₃ [1,25(OH)₂D₃] [69]. 1,25(OH)₂D₃ and low phosphate intake elevate serum phosphate levels by upregulating NaPi-IIb expression, while PTH reduces serum phosphate levels by promoting NaPi-IIa protein degradation and increasing phosphate excretion.
The KL gene plays a critical role in phosphate homeostasis [70]. Although KL is expressed in multiple tissues, its expression levels are highest in the kidneys, parathyroid glands, and choroid plexus. Initially identified as an anti-aging gene, KL deficiency leads to multi-organ dysfunction, including soft tissue calcification and osteomalacia [71]. Recent studies increasingly focus on the association between KL and nephrolithiasis formation [72, 74].
Von Kossa (VK) staining of KL–/– mice reveals extensive renal crystalline deposits, and renal proximal/distal tubules lose differentiation capacity [72], promoting Randall’s plaque formation. Zewu Zhu et al. discovered [74] KL downregulation in Randall’s plaque tissue, negatively correlated with osteogenic markers (MSX2, Runx2, OCN). Human renal interstitial fibroblasts (hRIFs) exhibit stronger osteogenic potential than human proximal tubular epithelial (HK-2) cells, with more pronounced KL of hRIFs decline following osteogenic induction. In co-culture, KL released by HK-2 cells inhibited hRIFs’ osteogenic differentiation by suppressing the Wnt-β-catenin signaling pathway [74]. This establishes KL deficiency as a causative factor in Randall’s plaque formation.
Role of steroid hormones in phosphate metabolism
Clinical studies indicate that men undergoing androgen deprivation therapy (e.g., GnRH analog treatment) exhibit elevated serum phosphate levels and enhanced renal phosphate reabsorption [75], while hypogonadal men receiving androgen therapy show a significant reduction in serum phosphate [76]. A randomized controlled trial further found that androgen supplementation in elderly men lowers plasma phosphate levels (without affecting FGF23 or Klotho) [77]. This suggests androgens may promote phosphate excretion by inhibiting renal phosphate reabsorption, potentially alleviating hyperphosphatemia and thereby reducing CaP stone formation, though this remains controversial.
The Rotterdam Study indicated that serum testosterone may partially explain gender differences in serum phosphate, while estradiol may contribute to gender differences in serum calcium [78]. Postmenopausal women experience a sudden decline in estrogen, leading to elevated serum calcium, phosphate concentrations, and calcium-phosphate product [79], thereby increasing the risk of nephrolithiasis. Women receiving estrogen therapy exhibit reduced serum phosphate levels, as estrogen inhibits NaPi co-transporter in renal proximal tubules [80]. In vivo and vitro studies confirm that rat proximal tubule (PT) cells express both ERα and ERβ, and 17β-estradiol (17β-E_2_) induces phosphaturia by directly and specifically targeting NaPi-IIa in PT cells [81, 82]. Estrogen also causes phosphaturia and hypophosphatemia in mice by downregulating NaPi-IIa and NaPi-IIc proteins in proximal tubules via ERα activation. And, this is independent of PTH levels [83].
Serum phosphate levels decrease with age in both genders [84]. Estrogen reduces phosphate excretion by downregulating NaPi-IIa (studies on androgen effects on NaPi-IIa are relatively scarce), NaPi-IIa downregulation is identified as a risk factor in renal calculi research. Notably, both human and animal studies indicate that due to higher urinary pH in females [85], young females (with normal estrogen secretion) are more prone to CaP crystal formation, while males are more susceptible to CaOx crystals [86, 87]. Disrupted phosphate levels clearly contribute to nephrolithiasis formation or renal tissue damage. Nevertheless, the relationship between steroid hormones, phosphate homeostasis, and Randall’s plaque formation requires more comprehensive research to validate.
A cross-sectional study reported that a significant association between KL and steroid hormones in the U.S. male population. KL levels increased with total testosterone, estradiol and sex hormone-binding globulin levels [88]. However, male testosterone supplementation does not affect KL levels [77]. In response to mild phosphate challenges, females (especially aged females) maintain normal serum phosphorus through significant upregulation of FGF23, forming a protective mechanism of compensatory FGF23 elevation [89]. Males, conversely, rely on maintaining KL expression to ensure FGF23 signaling efficiency, yet their serum FGF23 levels are markedly lower than females (approximately 50% in aged males with high phosphorus compared to aged females with high phosphorus) [89]. This disparity suggests that despite higher KL levels in males, insufficient FGF23 substrate may compromise the long-term stability of FGF23-KL signaling. In contrast, females’ compensatory elevation of FGF23 enables a more proactive response to phosphorus load, reducing renal phosphorus reabsorption pressure and indirectly lowering the risk of renal injury [90]. Therefore, variations in KL expression may contribute to the greater severity of renal injury in males compared to females during progressive kidney disease [91]. Based on this, it can be inferred that estrogen may maintain serum Phosphate levels within the normal range via KL, thereby preventing mineral metabolism disorders.
Notably, in the proximal tubule epithelial cells of KL–/– mice, NaPi-IIa cotransporter expression is elevated, leading to hyperphosphatemia, which an effect reversed by dual knockout of NaPi-IIa and KL mice. Double knockout mice also exhibit reversed aging characteristics, suppressed renal calcification, and significantly extended lifespan, but high-phosphate diets restore premature aging phenotypes [92]. This confirms that phosphate toxicity is a primary cause of premature aging, inducing renal tubular epithelial cell death and other cytotoxic effects that disrupt mineral metabolism and exacerbate chronic kidney disease [93, 94].
In summary, for nephrolithiasis patients, potential mechanisms of estrogen therapy may include: (i) downregulating NaPi-IIa expression to inhibit phosphate toxicity from hyperphosphatemia [82], (ii) upregulating KL expression to prevent cellular/tissue senescence and death, and inhibiting osteogenic differentiation of hRIFs [74]. While this strategy has not been applied to nephrolithiasis treatment, similar mechanisms have been validated in estrogen therapy for acute heart failure [95]. Their experiment demonstrated that 17β-estradiol treatment suppressed adrenal NaPi-IIa expression, normalizing serum phosphorus levels and reversing cardiac mitochondrial respiratory enzyme dysfunction, excessive ROS production, oxidative stress, and cardiomyocyte apoptosis in KL–/– mice. Given the strong association between nephrolithiasis and cardiovascular disease [96, 97], estrogen’s potential role in treating nephrolithiasis via this mechanism merits further in-depth investigation (Fig. 3).
Fig. 3. Role of steroid hormones in phosphate and calcium metabolism (A) Phosphate transport in the proximal renal tubule. Estrogen (red arrow) reduces NaPi-IIa transport, inhibiting phosphate excretion. (B) Calcium transport in the distal convoluted tubule and collecting duct. Estrogen (red arrow) enhances transport via the TPRV5-Calbindin-PMCA/NCX1 pathway, increasing calcium reabsorption and preventing hypercalciuria. Androgen (blue arrow) has the opposite effect, which role in PMCA remains uncertain. NaPi-IIa, sodium- dependent phosphate transport protein IIa; NaPi-IIc, sodium- dependent phosphate transport protein IIc; TRPV5, transient receptor potential vanilloid channel 5; PMCA, plasma membrane calcium-ATPase; NCX1, sodium-calcium exchanger 1
Steroid hormones and calcium-containing stones: regulation of calcium metabolism
Calcium metabolism and nephrolithiasis formation
Renal calcium reabsorption via two pathways, with dysfunction in either disrupting calcium balance and increasing stones risk [98]. Paracellular passive transport primarily occurs in the proximal tubule (PT, 60–70%) and the thick ascending limb of the loop of Henle (TAL, 20%) [99].
Transcellular active transport occurs primarily in the distal convoluted tubule (DT, 10%) and collecting duct (CT, 5%) [100] (Fig. 3). Calcium influx across the apical membrane of DT cells is mediated by the transient receptor potential vanilloid channel 5 (TRPV5), followed by intracellular transport via Calbindin-D28k (in DT principal cells) or Calbindin-D9k (in connecting tubules). Finally, calcium is transported into the bloodstream via plasma membrane calcium-ATPase (PMCA) and sodium-calcium exchanger 1 (NCX1) in the basolateral membrane [101].
Dysfunction of the TRPV5-Calbindin-PMCA1b transport pathway is directly implicated in idiopathic hypercalciuria kidney stones disease [102, 103]. TRPV5–/– mice confirm that TRPV5 is a key regulator of renal tubular Ca²⁺ reabsorption [104], altered TRPV5 expression disrupts pathway integrity, leading to tubular calcium deposition and stone formation. Abnormal TRPV5 expression or function is also directly associated with increased recurrence risk in calcium-containing nephrolithiasis patients [105].
1,25(OH)₂D₃ and PTH also participate in calcium homeostasis regulation [36, 106]. 1,25(OH)₂D₃ is a key mediator of vitamin D’s effects on calcium and bone metabolism [107]. In clinical trials, multivariate regression analysis demonstrated a positive correlation between 1,25(OH)₂D₃ and urinary calcium excretion [108]. Shakhssalim et al. identified 1,25(OH)₂D₃ as a key hormone in recurrent nephrolithiasis pathogenesis, increasing stone risk by increasing urinary calcium excretion [109]. In both humans and rats, vitamin D supplementation has been shown to cause hypercalciuria, nephrolithiasis, and/or nephrocalcinosis [110, 111]. Vitamin D deficiency (defined as 25(OH)D < 20 ng/mL) is significantly more prevalent in nephrolithiasis patients than in non-stone patients, and may elevate PTH levels, further increasing stone risk [112, 113]. PTH converts 25-hydroxyvitamin D[25(OH)D] to active 1,25(OH)₂D₃ to promote intestinal and renal calcium absorption, and stimulates bone resorption to raise serum calcium [114]. Overactivation of PTH’s calcium-raising effects leads to accelerated bone resorption (elevating serum calcium) and inhibited renal calcium reabsorption (increasing urinary calcium), promoting crystal formation with oxalate and phosphate, and ultimately stone development [108, 115, 116]. Clinically, patients with primary hyperparathyroidism (excessive PTH secretion) exhibit a significantly increased incidence of nephrolithiasis [34, 117, 119].
Role of steroid hormones in calcium metabolism
Steroid hormones (estrogens, androgens) regulate renal calcium transport through direct or indirect pathways (Fig. 3), though results remain controversial due to experimental heterogeneity (e.g., calcium loading, species, model types), making calcium metabolism a key focus of current research.
Mouse models lacking aromatase (with impaired estrogen synthesis) exhibit hypercalciuria and significantly reduced expression of renal transporters TRPV5, PMCA1b, NCX1, and Calbindin-D28k. Treatment with E_2_ fully restored the function of transporters including TRPV5 [120]. Following ovariectomy in mature rats, estradiol (E_2_) supplementation at varying doses increases serum calcium, reduces urinary calcium excretion, and upregulates renal transporters including TRPV5, with no significant changes in protein levels following 1,25(OH)₂-vitamin D₃ addition, indicating this process is independent of the vitamin D pathway [121]. These findings demonstrate that estrogen directly stabilizes Ca²⁺ homeostasis by regulating calcium transporters in DT and CT, inhibiting nephrolithiasis development.
Estrogen-deficient female rats fed low-calcium diets exhibit negative calcium balance, accompanied by downregulation of TRPV5, CaBP28k, and PMCA1b, increasing stone risk [122]. In contrast, ovariectomized rats on high-calcium diets show positive calcium balance, suggesting that estrogen deficient-mediated regulation of calcium metabolism may be influenced by calcium intake levels [122].
A randomized controlled trial of 36,282 postmenopausal women found that the intervention group (500 mg calcium carbonate + 200 IU vitamin D₃ twice daily, achieving an average daily calcium intake of 2100 mg) had a 17% higher risk of nephrolithiasis than the placebo group after 7 years of follow-up [123]. An observational analysis from the Nurses’ Health Study I (NHS I) reached similar conclusions: women taking calcium supplements had a 20% increased risk of stone events [124]. However, data from the Health Professionals Follow-up Study (45,619 healthy men without stone history) showed that the low-calcium diet group (797 ± 85 mg/day) had a 50% lower stone risk than the normal intake group (1307 ± 280 mg/day) [125]. Another randomized controlled trial in patients with recurrent calcium oxalate stones and hypercalciuria confirmed that the balanced calcium group (1200 mg/day) exhibited significantly reduced urinary oxalate excretion and a 50% lower risk of new stones after 5 years, while the ultra-low calcium group (400 mg/day) showed higher urinary oxalate excretion [126]. Collectively, balanced calcium intake has become a core component of dietary recommendations for nephrolithiasis patients, as it reduces stone formation risk by maintaining calcium metabolic balance.
Postmenopausal women face increased risks of fractures and osteoporosis, primarily due to uncontrolled osteoclast activity (bone resorption exceeding formation), reduced vitamin D activation, and diminished calcium reabsorption efficiency, leading to rapid bone loss, hypercalciuria, and elevated nephrolithiasis risk [127, 128]. The NHS I confirmed that both natural and surgical menopause are associated with higher nephrolithiasis incidence [129]. In vitamin-deficient mice, reduced testosterone and estrogen levels have also been observed [130]. However, the Women’s Health Initiative (WHI) clinical trial found that vitamin D and calcium supplementation inhibited fractures but increased nephrolithiasis risk [123]. In women receiving estrogen therapy, elevated 1,25(OH)₂D₃ levels reduced urinary calcium excretion, decreasing hypercalciuria incidence [131]. A network meta-analysis exploring the efficacy of drug therapies in preventing fractures in postmenopausal women noted that estrogen reduces fracture incidence [132].Therefor, maybe prioritizing estrogen for treating osteoporosis or nephrolithiasis in postmenopausal women, as it elevates 1,25(OH)₂D₃ levels without the side effects of calcium/vitamin D supplementation. However, estrogen dosage must be carefully managed. Tissue-selective estrogen complexes (TSECs) [133], combining selective estrogen receptor modulators (SERMs) with one or more estrogens, have been used for fracture/osteoporosis treatment, offering better tolerance and safety than monotherapy by exerting neutral or antagonistic effects on tissues where estrogen action is undesirable (breast and endometrium) [134].
While urinary calcium excretion is consistently higher in males than females, men with low testosterone levels exhibit a lower risk of nephrolithiasis [50]. Immunohistochemical studies have linked nephrolithiasis to upregulated AR expression in renal tissue [135], and AR–/– mice show significantly reduced calcium oxalate crystal formation [59]. A cross-sectional study further demonstrated a positive correlation between serum testosterone levels and urinary calcium excretion in males, suggesting androgens may directly inhibit renal Ca²⁺ reabsorption via AR, increasing urinary Ca²⁺ saturation and stone risk [136].
In vitro experiments showed that dihydrotestosterone (DHT) treatment of rabbit DCT-CT cells inhibited apical-to-basolateral calcium transport (PMCA) [137]. In contrast, immortalized DT cells incubated with T and DHT for 24 h exhibited increased PMCA activity, enhancing calcium uptake [138]. While T is the primary circulating androgen in male, exerting effects via the AR by converting (through 5α-reductase) to DHT or via ER (following aromatization to estradiol) [139]. DHT possesses 2–3 times the biological activity of T and exhibits higher affinity for target tissues [140]. Additionally, animal studies suggest finasteride (a 5α-reductase inhibitor) may be used to treat nephrolithiasis [141]. Animal studies have yielded conflicting results. Hypercalciuria induced in male rats 2 [142] and 8 weeks [143] after orchidectomy was reversed by treatment with either T or DHT. another study found that castration in male mice reduced urinary calcium excretion and increased expression of transporters like TRPV5, while T supplementation increased urinary calcium excretion and downregulated these transporters [137]. Rougin Khalil et al. found that castrated rats develop hypercalciuria, which is prevented by bisphosphonates (inhibitors of bone resorption). Moreover, combining bisphosphonate treatment with an extremely low-calcium diet did not reduce urinary calcium levels post-castration. This suggests androgens may interact dynamically with renal, skeletal, and intestinal calcium homeostasis [144].
Androgens may indirectly regulate calcium transport by influencing vitamin D metabolism. Therefore, in the presence of androgens in PT, the synthesis of 1,25(OH)₂D₃ is inhibited. This leads to impaired active phosphate reabsorption at the proximal tubule level while simultaneously suppressing active calcium reabsorption at DT level, triggering hypercalciuria and other metabolic abnormalities [144]. In pregnancy, hyperandrogenism may adversely affect placental vitamin D metabolism. Studies reveal testosterone significantly inhibits CYP27B1 (which synthetic vitamin D) while stimulating CYP24A1 (which degrades vitamin D) expression in cultured trophoblasts [145].
In summary, estrogen tends to reduce urinary calcium by enhancing renal calcium reabsorption, decreasing bone calcium loss, and upregulating calcium transporters. However, the effects of androgens on calcium metabolism remain contradictory. First, calcium homeostasis results from the coordinated actions of the kidneys, bones, and intestines. Second, within tissues, T can act as either T or DHT via the AR, or as estradiol via the ER. Further rigorous research is needed to clarify the precise role of steroid hormones in calcium metabolism and nephrolithiasis formation.
Steroid hormones and nephrolithiasis: regulation of inflammation
Nephrolithiasis formation and inflammatory responses exist in a vicious cycle of bidirectional regulation [146]. After calcium oxalate and calcium phosphate crystals precipitate, they damage renal tubular epithelial cells and activate inflammatory pathways, while inflammation further exacerbates crystal deposition and tissue injury [8, 147, 148]. steroid hormones exert effects on this inflammatory process through receptor-mediated pathway regulation, with estrogen exhibiting anti-inflammatory properties and androgens promoting inflammation, thereby further influencing stone progression (Fig. 4).
Inflammation and stones formation and growth
When calcium oxalate, calcium phosphate, and other crystals become supersaturated and precipitate in urine, the positive charge on their surface interacts with the negative charge of phospholipids on renal tubular epithelial cell membranes, disrupting membrane integrity and releasing damage-associated molecular patterns (DAMPs) such as ATP and HMGB1 [149]. This interaction also exposes annexin A1 [150] and α-enolase [151], enhancing crystal-cell adhesion. Additionally, crystals activate NADPH oxidase within tubular epithelial cells (e.g., p22-phox, NOX4 subunits), promoting reactive oxygen species (ROS) production [152, 153].
In nephrolithiasis pathophysiology, NADPH oxidase is the primary renal source of ROS, while mitochondrial ROS (mtROS) is a significant secondary contributor to ROS-related renal injury [153]. ROS not only directly oxidatively damage cell membranes but also activates the TXNIP-NLRP3 pathway, generating mature pro-inflammatory factors (such as IL-1β, IL-8, and IL-10) that are secreted extracellularly, recruiting immune cells like monocytes and neutrophils to infiltrate the renal interstitium [154, 155]. Furthermore, impaired clearance of damaged mitochondria (due to autophagy dysfunction) releases mtDNA and cytochrome C (initiating apoptosis), exacerbating cell detachment and dysfunction [156, 157]. Crystal stimulation or factors like IL-1β can activate NF-κB inflammatory pathways, upregulating expression of pro-inflammatory genes including TNF-α, IL-6, and MCP-1 (monocyte chemotactic protein-1), further amplifying inflammatory infiltration [158, 159].
Role of steroid hormones in inflammation
Estrogens exert their effects primarily through two nuclear receptors (ERα, ERβ) and a membrane receptor (GPER) [160]. ERβ is highly expressed in renal tissues (renal tubular epithelial cells, macrophages) and serves as a key anti-inflammatory receptor [161, 162]. Upon binding to ERβ, estrogens directly regulate the transcription of anti-inflammatory genes (such as IL-10, TGF-β) or suppress pro-inflammatory gene expression by interacting with other transcription factors (e.g., AP-1) [163, 164]. Estrogen also protects against acute kidney injury via ERα, though ERα has weaker direct pathological relevance to stone formation than ERβ [165]. In hyperoxaluria-induced ERβ−/−mice, renal IL-1β and TNF-α levels are significantly increased, with calcium oxalate deposition reaching 2–3 times higher than in wild-type mice. While ERα−/− mice exhibit only mildly abnormal inflammatory markers, which could be improved through ERβ compensation [60]. Thus, ERβ plays a dominant anti-inflammatory role in the kidney, while ERα is secondary. GPER also participates in inflammatory responses. The GPER1 agonist G-1 protect human renal tubular epithelial cells from damage [166].
Estrogen reduces renal inflammatory damage by upregulating antioxidant enzyme activity (SOD1, CAT) in renal tissue, decreasing ROS production [167, 168]. In vitro studies revealed [169] that E_2_ treatment reduced the release of annexin A1 and α-enolase from renal tubular epithelial cells under hyperoxalic acid conditions, decreases TLR4/NF-κB activity, and ultimately diminishes IL-1β and IL-18 release, inhibiting crystal adhesion to tubular epithelial cells and enhancing cell proliferation and tissue healing [151]. Postmenopausal women exhibit a twofold increase in nephrolithiasis incidence, accompanied by markedly elevated urinary inflammatory markers [50].
In contrast, androgens are key factors inducing renal injury. AR are widely distributed in renal tubular epithelial cells, renal interstitial macrophages, and hepatocytes [170]. Studies in AR−/− rat models confirm that AR is associated with upregulation of nephritis markers, cell death, and fibrosis markers [171]. Liang et al. discovered that androgen signaling pathways directly upregulate hepatic GO and the renal epithelial NADPH subunit P22-PHOx at the transcriptional level, potentially enhancing oxalate biosynthesis and leading to nephrolithiasis [59]. Androgens also increase surface α-enolase expression, enhancing calcium oxalate crystal adhesion to the apical membrane of renal tubular epithelial cells [172]. Another newly developed androgen receptor degradation enhancer, dimethylcurcumin (ASC-J9), has been shown to inhibit oxalate crystal formation by modulating oxalate biosynthesis and ROS-induced injury in rat renal tubular epithelial cells [173, 174]. Proteomics analysis of individuals receiving sex steroid hormone therapy (estradiol, testosterone) revealed that proteins involved in endothelial function (SFRP4, SOD3), anti-inflammation (TSG-6), and renal tissue structure maintenance are positively correlated with estradiol but negatively correlated with testosterone. Additionally, testosterone elevates levels of tubular injury biomarkers (YKL-40, MCP-1), further confirming estrogen’s protective role for tubular epithelial cells, while androgens exert the opposite effect [175].
Mitochondria, cellular powerhouses and major ROS sources, play a pivotal role in initiating kidney stone-related inflammation, crystal adhesion, and cellular damage-induced apoptosis [176]. Estrogen exerts mitochondrial protective effects via ER by upregulating mitochondrial autophagy-related proteins to promote clearance of damaged mitochondria and enhancing mitochondrial antioxidant capacity [177]. In contrast, androgens exacerbate mitochondrial dysfunction via AR. SIRT3, a nicotinamide adenine dinucleotide (NAD)-dependent class III histone deacetylase, functions within mitochondria to enhance respiration and reduce ROS production [178]. In acute kidney injury models, sex steroid hormone-dependent differences in renal mitochondrial Sirt3 (mtSirt3) expression have been observed that estradiol increases mtSirt3 protein and testosterone decreases it [179]. BNIP3, a gene mediating necrotic-like cell death by inducing mitochondrial permeability transition pore opening and mitochondrial dysfunction [180, 181]. In human and mouse renal tubular epithelial cell (RTEC) assays, mitochondrial membrane potential (ΔΨm) was significantly higher in the testosterone-treated group than in the normal control group, while BNIP3 siRNA treatment reduced ΔΨm compared to control and negative control siRNA groups [182]. Thus, testosterone induces apoptosis via BNIP3-mediated mitochondrial dysfunction, and RTEC death represents a key pathophysiological process in renal stone development [158]. And, testosterone promotes apoptotic damage in human renal tubular cells [183]. Sex steroid hormone-mediated differential regulation of mitochondria and related genes may serve as potential therapeutic targets to mitigate excessive ROS-induced renal injury and apoptotic shedding.
As previously described, disproportionate calcium oxalate exposure activates the NF-κB pathway, triggering the production of numerous inflammatory cytokines [184] that damage mitochondria (generating ROS) and induce macrophage recruitment, depending on cytokine concentration, exposure duration, and cytokine competition [154]. M1 macrophages promote renal crystal development, while M2 macrophages reduce pro-inflammatory factor expression through crystal phagocytosis and inhibit stone formation [185, 186].
Studies using mouse macrophages (RAW264.7 and female murine bone marrow macrophages) show that 24-hour E_2_ treatment upregulates Sirt3 and attenuates oxidative stress and pro-inflammatory polarization in M1 macrophages [187]. Estrogen also upregulates CSF-1 (colony-stimulating factor 1) receptor expression on macrophage surfaces, enhancing CSF-1-mediated M2 polarization signaling [188]. M2 induce secretion of anti-inflammatory factors such as IL-10 and TGF-β, and enhancing macrophage phagocytic capacity toward calcium oxalate crystals [189]. Androgens promote M1 macrophage activation via AR while inhibiting M2 polarization, disrupting the renal inflammatory microenvironment balance and suppressing intrarenal macrophage phagocytosis of CaOx crystals [188]. Mechanistic analysis indicates that AR reduces macrophage CSF-1 expression via increased miRNA-185-5p expression, inhibiting M2 macrophage polarization-mediated phagocytosis of CaOx crystals [174]. A prospective study further found that CaOx renal stones are positively correlated with genetic expression of AR and miRNA, and inversely related to CSF-1 [190]. Collectively, immunoregulation of inflammatory cytokines offers novel insights into macrophage-mediated prevention and treatment of nephrolithiasis, with inflammatory cytokines potentially representing a key pathway through which steroid hormones influence macrophage polarization in nephrolithiasis disease.
Existing research indicates a close relationship between vascular calcification and nephrolithiasis, with vascular calcification accelerating the stone progression, while nephrolithiasis also often accompanied by vascular calcification [190, 192]. Osteopontin (OPN) and matrix Gla protein (MGP) are two crucial calcification inhibitors in the human body, preventing both vascular calcification and nephrolithiasis [193, 194]. Animal studies indicate that compared to mice with normal OPN expression, high-phosphate-fed OPN−/− mice exhibit more severe vascular calcification and renal calcification, suggesting OPN can simultaneously prevent nephrocalcinosis and vascular calcification [195]. Studies on Sprague-Dawley rats before and after castration indicate that estrogen inhibit stone formation by increasing renal OPN expression and reducing urinary oxalate excretion, while androgens exert the opposite effect [18]. Miyajima et al. also demonstrated in a hyperoxaluric rat model that exogenous estrogen administration to the ovariectomized (OVX) rat increased OPN mRNA expression, consistent with results from primary cultured rat renal cells. Furthermore, ERα protected renal epithelial cells from damage in the presence of estrogen [196]. In vascular calcification, estrogen maintains vascular smooth muscle cell (VSMC) functional integrity by promoting OPN expression via G protein-coupled receptor 30 (GPR30) [197]. MGP is the most potent inhibitor of tissue calcification [194, 198]. MGP-deficient mice spontaneously develop arterial calcification [199]. Numerous studies have linked MGP gene polymorphisms to increased nephrolithiasis susceptibility [200, 201]. Estrogen also increase MGP expression, slowing phosphate-induced osteogenic differentiation of VSMCs [202], delaying the progression of nephrolithiasis.
In summary, estrogen suppresses nephrolithiasis-related inflammation through ER anti-inflammatory pathways, mitochondrial protection, and M2 macrophage polarization, whereas androgens exert the opposite effect (Fig. 4).
Fig. 4. Role of steroid hormones in Inflammation (a) Hyperoxaluria, hypercalciuria and hypocitraturia are risk factors involved in the pathogenesis of nephrolithiasis and can lead to epithelial cell damage. Androgens (Blue, b) bind to the androgen receptor (AR), increasing α-enolase and promoting adhesion between crystals and cells (c). Estrogens (Red, d) bind to the estrogen receptor (ER), inhibiting adhesion between crystals and cells (e). NADPH is a key contributor to ROS-induced inflammation, promoted by androgens (f). Estrogen inhibits the production of ROS (g). Mitochondria are also major ROS sources (h). Androgen impair mitochondrial function by increasing BNIP3 and ΔΨm, elevating ROS and accelerating apoptosis (i). Conversely, estrogens enhance Sirt3 to maintain mitochondrial function (j). Inflammatory factors produced during inflammation recruit macrophages (k). Estrogen promotes anti-inflammatory M2 macrophages to phagocytose crystals via CSF-1 (l), while androgens suppress M2 by increasing miRNA-185-5p expression, which reduces CSF-1 expression and increases pro-inflammatory M1 macrophages (m). The increased pro-inflammatory macrophages further sustain the inflammatory microenvironment, promoting crystal nucleation and adhesion in a self-perpetuating cycle**(n)**. Pro-inflammatory macrophages, mitochondrial damage, and the accompanying inflammation drive collagen deposition and mineralization (o), ultimately leading to nephrolithiasis formation. ΔΨm, mitochondrial membrane potential; CSF-1, colony-stimulating factor 1
Clinical prospects and challenges of sex hormone therapy for nephrolithiasis
A clinical cross-sectional study employing a questionnaire survey finds that natural and surgical menopause are associated with nephrolithiasis, while postmenopausal estrogen therapy showed no association with stone risk in postmenopausal women [131]. Similarly, in 91,731 female Nurses’ Health Study participants, estrogen use was unrelated to nephrolithiasis occurrence in postmenopausal women [203]. However, retrospective studies suggest that estrogen therapy in postmenopausal women with nephrolithiasis may reduce recurrence risk by lowering urinary calcium and calcium oxalate saturation [204]. Despite this potential benefit, estrogen replacement therapy is limited as a routine preventive measure for nephrolithiasis due to increased risks of thrombosis and breast cancer [205]. The combined use of estrogen and selective estrogen receptor modulators (SERMs) may offer a promising alternative for treating nephrolithiasis while avoiding some side effects, though this approach has not been explored in nephrolithiasis-related studies [133, 206, 207]. It has, however, been applied to the treatment of other related kidney diseases (such as renal ischemia-reperfusion injury and chronic kidney disease) [165, 208, 209]. Developing strategies that provide renal protection while avoiding systemic side effects represents an important future research direction.
Given androgens’ active role in promoting stone-forming substance excretion and renal inflammation, antagonizing the AR signaling pathway theoretically serves as a strategy for preventing and treating male nephrolithiasis. However, systemic AR antagonists (such as bicalutamide) are associated with side effects such as decreased libido and gynecomastia, limiting their use exclusively for stone prevention [210, 212]. Finasteride (a 5α-reductase inhibitor) inhibits testosterone-induced calcium oxalate crystallization and crystal-cell adhesion [141]. Novel AR degradation promoters like ASC-J9 show promising efficacy in kidney disease [59, 173]. ASC-J9 suppresses renal cell carcinoma progression by targeting an AR-dependent HIF-α/VEGF signaling pathway [213] and may reduce intrarenal CaOx crystal deposition by altering macrophage recruitment/M2 polarization [174]. However, its safety and efficacy require validation through human clinical trials.
Phytoestrogens, natural plant-derived compounds with a structure resembling estradiol, can bind to ER and exert estrogenic effects [214, 216]. Daidzein, a soy isoflavones, exerts pharmacological effects by binding to ERα and β [217]. In male menopausal rat models, daidzein upregulates the anti-aging protein KL and the NaPi-IIa cotransporter, stabilizing calcium and phosphate homeostasis. Notably, unlike estrogen therapy (which reduces NaPi-IIa levels), daidzein may exert rapid non-genomic effects by activating G-protein-coupled receptor 30 (GPR30/GPER) [218], while estradiol treatment activates the mitogen-activated protein kinase (MEK 1/2) [219]. Trigonelline, the second most abundant bioactive alkaloid in coffee, is also classified as a phytoestrogen. By inhibiting the formation, growth, and adhesion of calcium oxalate crystals to cells, while simultaneously downregulating crystal receptor expression, the formation of nephrolithiasis is suppressed [220]. Kaempferol, one of the most common flavonoids, inhibits AR expression, oxidative stress and inflammation by regulating the AR/NOX2 signaling pathway, reducing CaOx crystal deposition and crystal-induced kidney damage, and inhibiting stone formation [217].
In summary, based on the established core regulatory role of steroid hormones in nephrolithiasis formation, targeting sex hormone signaling pathways may emerge as a potential new strategy for prevention and treatment.
Conclusion
The prevalence of nephrolithiasis in women has risen significantly, narrowing the gender gap. Women today bear a heavier burden regarding obesity, weight-loss surgery, and dieting. Compared to men, women have higher urinary pH levels, leading to greater absorption of dietary organic anions, all factors associated with increased stone formation. However, research indicates that several differences in stone formation mechanisms persist between men and women. Increasing evidence confirms the influence of steroid hormones on stone formation and the associated inflammatory microenvironment. Furthermore, these gender differences in stones may offer new insights for developing personalized treatment strategies in the future.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bargagli M, Ferraro PM, Vittori M, Lombardi G, Gambaro G, Somani B. Calcium and Vitamin D supplementation and their association with kidney stone disease: A narrative review. Nutrients. 2021; 13(12):4363.10.3390/nu 13124363 PMC 870762734959915 · doi ↗ · pubmed ↗
- 2Letavernier E, Daudon M. Vitamin D, Hypercalciuria and Kidney Stones. Nutrients. 2018;10(3):366.10.3390/nu 10030366 PMC 587278429562593 · doi ↗ · pubmed ↗
- 3Van Abel M, Hoenderop JG, Dardenne O, St Arnaud R, Van Os CH, Van Leeuwen HJ, Bindels RJ, 1,25-dihydroxyvitamin D(3)-independent stimulatory effect of estrogen on the expression of E Ca C 1 in the kidney. J Am Soc Nephrol. 2002;13:2102–2109.10.1097/01.asn.0000022423.34922.2a 12138142 · doi ↗ · pubmed ↗
- 4van Eeghen SA, Pyle L, Narongkiatikhun P, Choi YJ, Obeid W, Parikh CR, Vosters TG, van Valkengoed IG, Krebber MM, Touw DJ, den Heijer M, Bjornstad P, van Raalte DH, Nokoff NJ. Unveiling mechanisms underlying kidney function changes during sex hormone therapy. J Clin Invest. 2025;135(9):e 190850.10.1172/JCI 190850 PMC 1204309540193283 · doi ↗ · pubmed ↗
- 5Juszczak F, Arnould T, Declèves AE. The role of mitochondrial sirtuins (SIRT 3, SIRT 4 and SIRT 5) in renal cell metabolism: Implication for kidney diseases. Int J Mol Sci. 2024;25(13):6936.10.3390/ijms 25136936 PMC 1124157039000044 · doi ↗ · pubmed ↗
- 6Barcena ML, Christiansen-Mensch C, Aslam M, Haritonow N, Ladilov Y, Regitz-Zagrosek V. Upregulation of mitochondrial sirt 3 and alleviation of the inflammatory phenotype in macrophages by estrogen. Cells. 2024;13(17):1420.10.3390/cells 13171420 PMC 1139387939272992 · doi ↗ · pubmed ↗
- 7Enright S, Werstuck GH. Investigating the effects of sex hormones on macrophage polarization. Int J Mol Sci. 2024;25(2):951.10.3390/ijms 25020951 PMC 1081617638256027 · doi ↗ · pubmed ↗
- 8Lee SJ, Lee IK, Jeon JH. Vascular calcification-new insights into its mechanism. Int J Mol Sci. 2020;21(8):2685.10.3390/ijms 21082685 PMC 721622832294899 · doi ↗ · pubmed ↗