Shared genetic architecture of psychoactive substance use and pan-cancer: insights from a large‑scale genome‑wide cross‑trait analysis
Jiahang Song, Pengzhu Li, Martin Canis, Kristian Unger, Nikolaus Alexander Haas, Olivier Gires

TL;DR
This study finds shared genetic links between psychoactive substance use and cancer, identifying potential drug targets that could help treat both conditions.
Contribution
The study reveals a shared genetic architecture between psychoactive substance use and cancer, identifying pleiotropic cerebellar hub genes and potential therapeutic targets.
Findings
34 shared trait pairs with significant genetic correlations between psychoactive substance use and cancer were identified.
21 cross-trait pleiotropic hub genes were found, expressed in the brain cerebellum.
CHRNA2, HRH3, and PTK6 were identified as potentially druggable targets after PheWAS analysis.
Abstract
Psychoactive substance use (PSU) and cancer are frequently observed comorbidities that have reciprocal influences and shared behavioral traits of the affected patients. While, e.g., nicotine and alcohol are major carcinogens in the etiology of lung and head and neck cancers, little is known about a shared overarching genetic architecture of PSU and cancer that may predispose individuals to both illnesses. Large-scale genome-wide association study (GWAS) summary data revealed shared genetic architecture between cancer and PSU, including alcohol use dependence (AlcUD) and nicotine use dependence (NicUD). Genetic correlations between PSU and cancer were assessed by linkage disequilibrium score regression (LDSC) and high-definition likelihood (HDL). Mendelian randomization (MR) analysis was additionally employed to explore causal associations between PSU and cancer. Moreover, phenome-wide…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
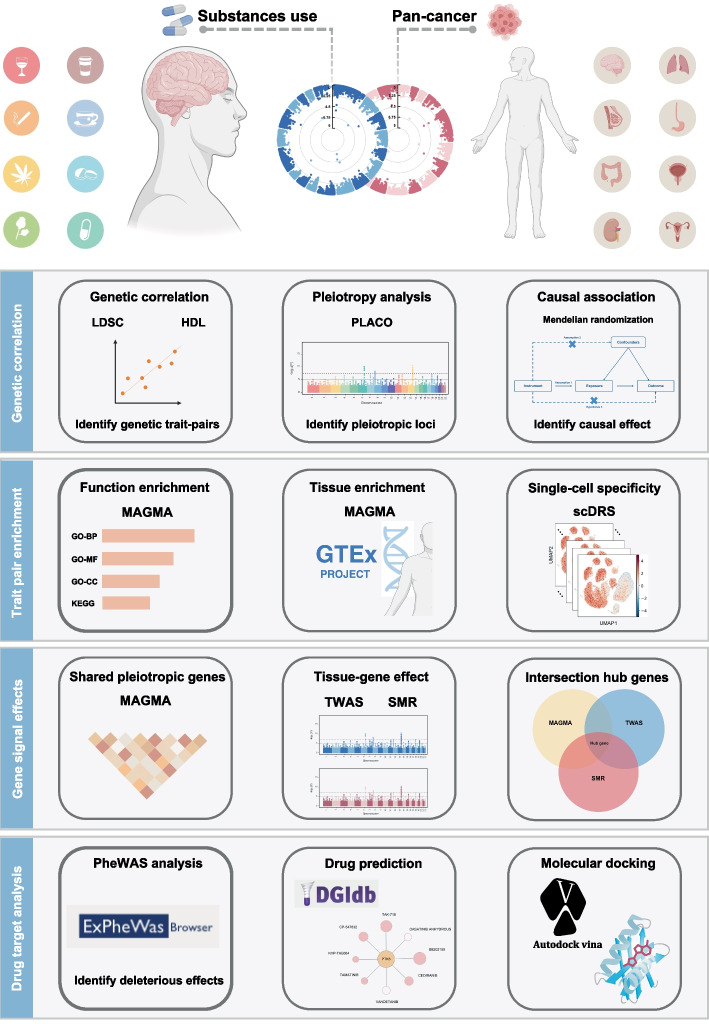 Figure 1
Figure 1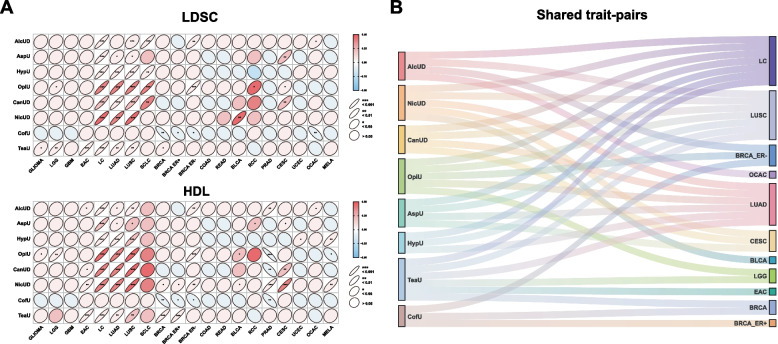 Figure 2
Figure 2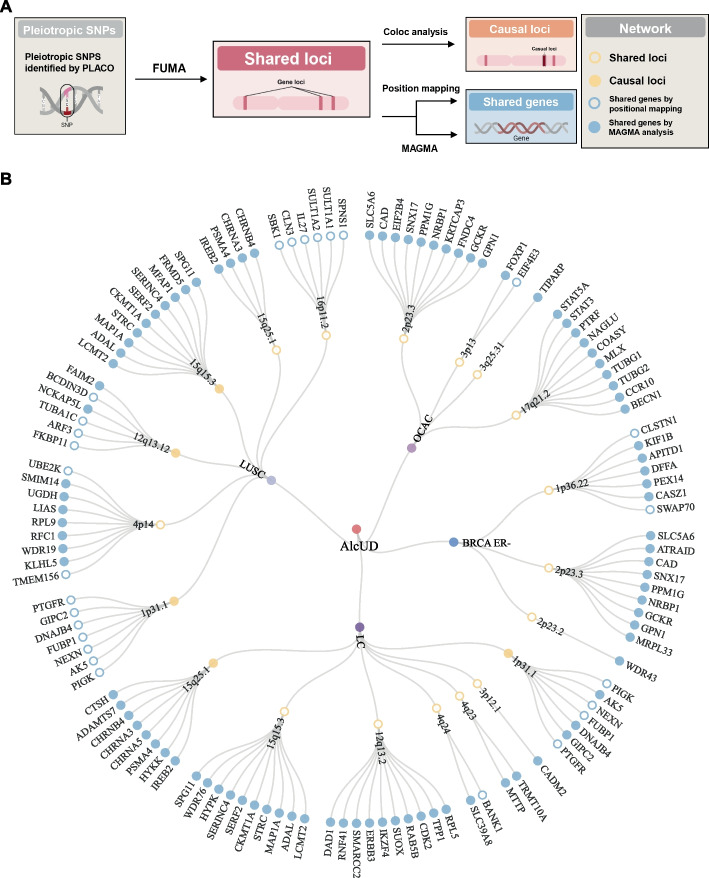 Figure 3
Figure 3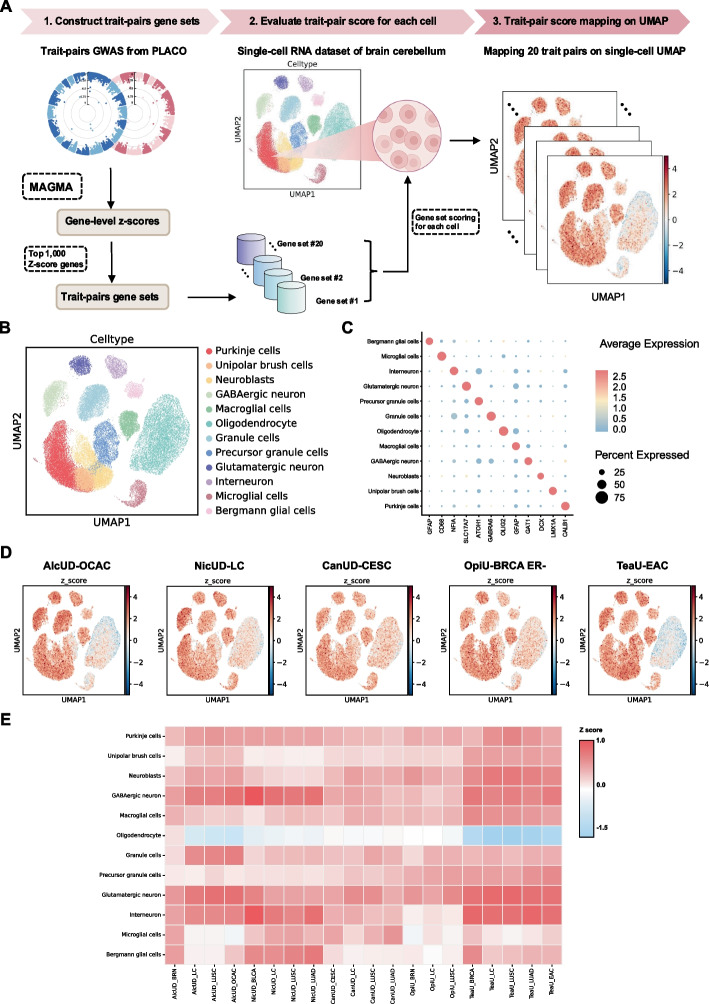 Figure 4
Figure 4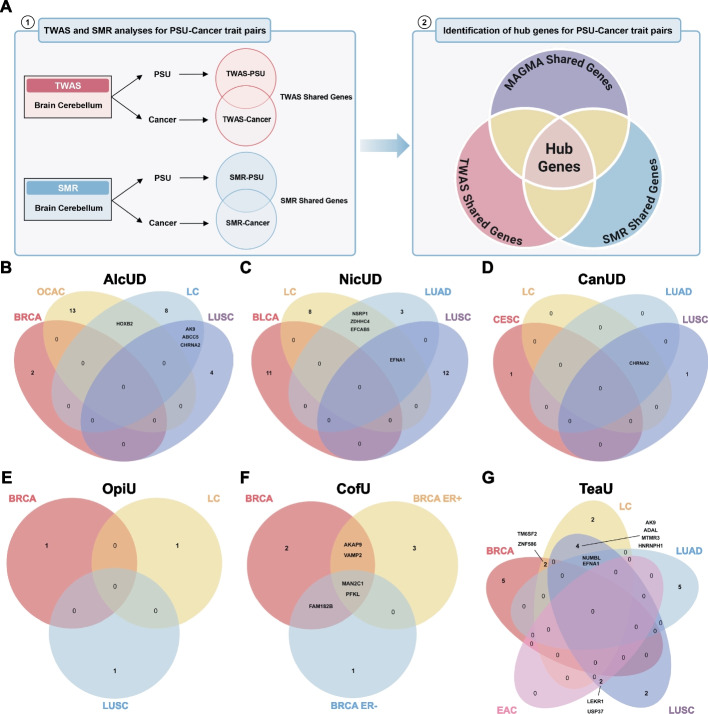 Figure 5
Figure 5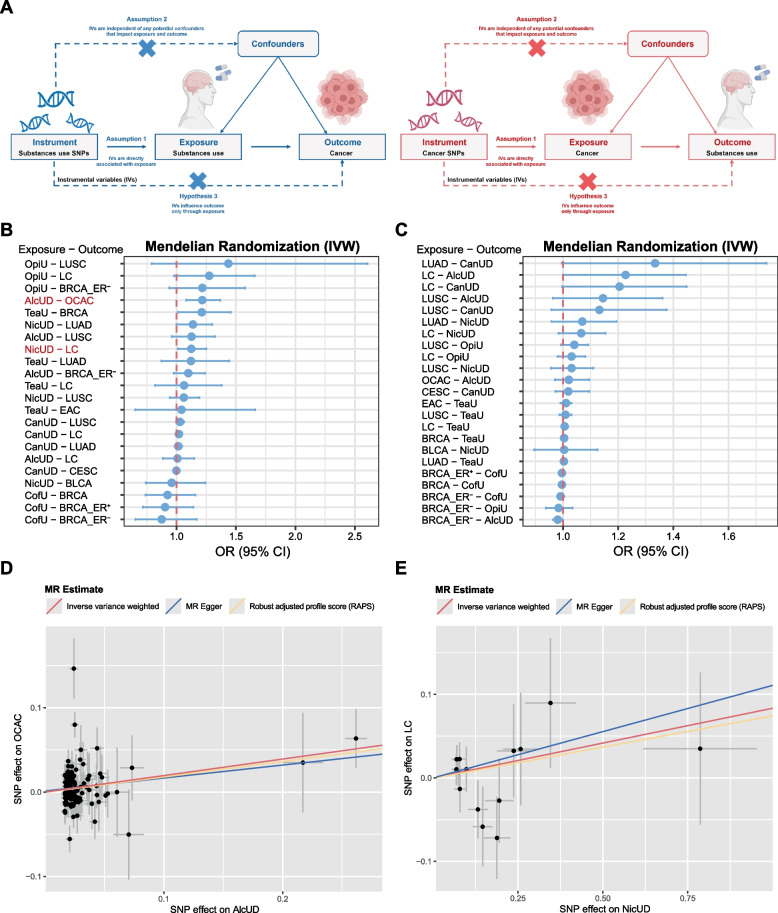 Figure 6
Figure 6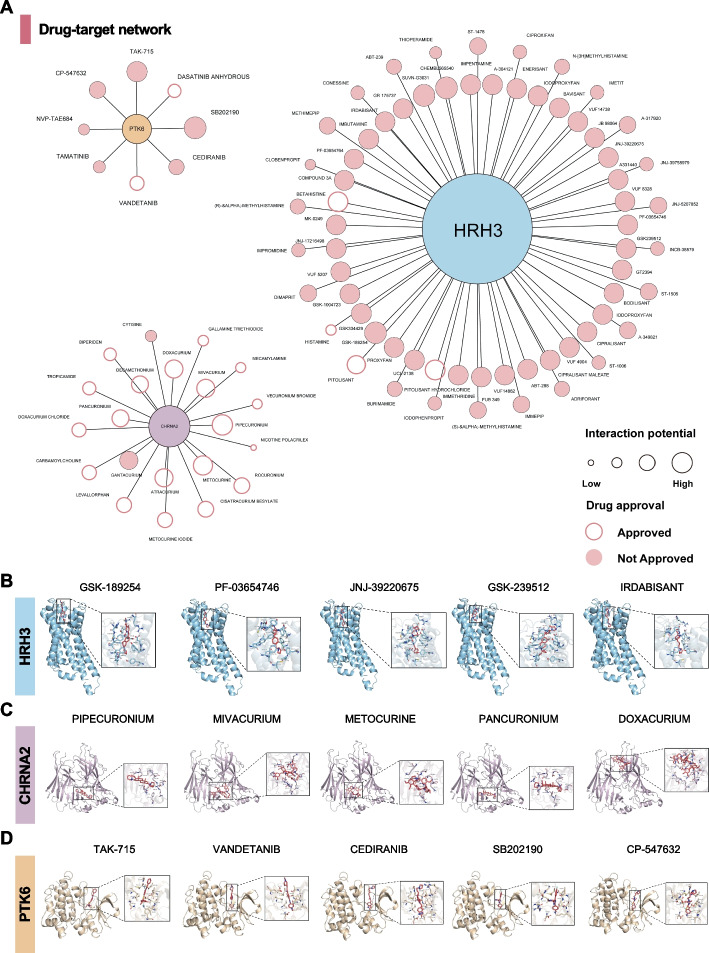 Figure 7
Figure 7- —https://doi.org/10.13039/501100010890Chinese Government Scholarship
- —Klinikum der Universität München (6933)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Associations and Epidemiology · Substance Abuse Treatment and Outcomes · Alcohol Consumption and Health Effects
Background
Substance use disorders (SUD) and psychoactive substance use (PSU) are very serious conditions that are associated with high relapse rates and morbidity. SUD/PSU have an additional health impact as they potentiate the development of illnesses including chronic pain, cardiovascular diseases, mental health disorders, and cancer [1–6]. In the following, we will refer to PSU as a converging term encompassing substance-related phenotypes such as use disorders or dependence and non-pathological use or consumption traits. The term cancer covers over 200 malignant diseases with pronounced inter- and intratumor molecular heterogeneity and variations in treatment response due to diverse underlying pathobiological, molecular mechanisms, and co-morbidities. PSU and tumorigenesis are both driven to varying degrees by genetic, epigenetic, environmental, and behavioral factors, and together cause major health and economic problems worldwide [1, 4, 7, 8].
Alcohol and nicotine dependence constitute major forms of PSU disorders with high global prevalence [9, 10]. Chronic and heavy consumption of alcohol and tobacco has been shown to greatly contribute to cancer development and progression [11–13]. For example, nicotine and alcohol abuse are major risk factors for head and neck squamous cell carcinoma [14–16]. In recent years, accumulating evidence has suggested shared genetic associations between alcohol dependence and cancers of the respiratory and digestive systems, as well as between nicotine dependence and lung cancer [17]. Cannabis is the most widely smoked substance after tobacco, and its prevalence continues to rise with the expansion of legal markets [18]. The increasing legalization and medical use of cannabis have prompted extensive research into its potential therapeutic effects on cancer-related symptoms such as pain, nausea, and loss of appetite, making it a possible adjunct therapy in cancer management [19, 20]. However, the association between cannabis use and cancer risk remains controversial. A large-scale cohort study reported that individuals with cannabis use disorder (CanUD) had more than a threefold increased risk of developing oral cancer compared with those without CanUD [21]. In contrast, some researchers have suggested a potential antitumor effect of cannabinoids, with evidence showing a significantly reduced risk of prostate cancer among individuals aged 50–64 who were current or former cannabis smokers [22]. Opioids, including prescription analgesics such as morphine, are widely used for pain management in cancer patients [23]. However, their extensive misuse and addictive potential have contributed to a global opioid crisis. Beyond their analgesic properties, opioids may regulate cancer progression through interactions with opioid receptors, which are closely associated with tumor growth and metastasis [24]. An observational study found that chronic opioid use among patients with persistent pain was associated with an increased risk of several cancers, including breast cancer [25]. Conversely, other studies have reported anti-tumor effects of opioids in leukemia [26]. Tea and coffee are among the most consumed beverages worldwide. Owing to their abundance of bioactive compounds such as catechins, tea polyphenols, caffeine, and various antioxidants [27, 28], researchers have shown great interest in their potential associations with cancer risk [29, 30]. A meta-analysis has suggested an inverse association between tea consumption and multiple types of cancer [31–33]. Similarly, some observational studies have suggested that coffee intake may reduce the risk of several cancers, including prostate, renal cell, and breast cancers [34–37], whereas other studies have reported conflicting findings [29, 38, 39]. However, traditional observational studies are inherently limited by residual confounding, selection bias, and measurement errors, which may obscure true relationships between PSU and cancer risk. Growing attention has been devoted to identifying common genetic foundations across diverse complex traits [40–42]. A previous study has revealed that SUD shares a common genetic structure with chronic pain [43]. Similarly, alcohol use disorder and body mass index exhibit genetic pleiotropy and shared neural associations [44]. Moreover, mood disorders and substance misuse have been shown to possess overlapping genetic architecture [45]. However, there is currently no systematic study exploring a potentially shared genetic architecture between PSU and cancer. Recent advances in genetic research have provided new perspectives on PSU and cancer, as genome-wide association studies (GWASs) have identified multiple genetic variants associated with each trait [46–51]. An underlying genetic architecture pre-dating sequential mutagenesis entailed by carcinogenic agents may account for an additional layer of heterogeneity [52]. The identification of such predispositions towards linked diseases rooted in population-wide genetic variations may help to better understand the course of life-threatening diseases. Therefore, we have conducted a large-scale cross-trait GWAS to identify common genetic architectures of PSU-cancer pairs in humans. We demonstrated the existence of shared loci and hub genes affected across trait pairs. MR methods further uncovered causal effects of PSU (AlcUD and NicUD) on cancer risk. These hub genes are primarily expressed in the brain cerebellum and encode druggable target candidates. Thus, we report on genetic architectures linking substance use and cancer development and identified common, actionable genes affected in various PSU-cancer trait pairs.
Methods
GWAS summary data source
(i) GWAS summary statistics for alcohol use dependence (AlcUD) [46] and cannabis use disorder (CanUD) [48] were obtained from the Psychiatric Genomics Consortium (PGC). Summary statistics for nicotine use dependence (NicUD) [53] were collected from the GWAS and Sequencing Consortium of Alcohol and Nicotine Use (GSCAN). Summary statistics for aspirin use (AspU) [54], opioid use (OpiU) [54], coffee use (CofU), and tea use (TeaU) were obtained from the UK Biobank [55]. Summary statistics for hypnotics use (HypU) were collected from the FinnGen R11 database [56]. In this study, PSU is used as an umbrella term encompassing a spectrum of substance-related phenotypes, ranging from use disorders or dependence (e.g., AlcUD, NicUD, CanUD) to non-pathological use or consumption traits (e.g., AspU, OpiU, HypU, CofU, and TeaU). Notably, not all PSU traits represent aberrant or disordered use.
(ii) GWAS summary statistics for 20 types of cancer were retrieved from public large-scale GWAS or GWAS meta-analyses: glioma (GLIOMA) [57], low-grade gliomas (LGG) [57], glioblastoma (GBM) [57], esophageal adenocarcinoma (EAC) [58, 59], lung cancer (LC) [50], lung adenocarcinoma (LUAD) [50], lung squamous cell carcinoma (LUSC) [50], small cell lung cancer (SCLC) [50], breast cancer (BRCA) [49], breast cancer with positive estrogen receptor (BRCA ER^+^) [49], breast cancer with negative estrogen receptor (BRCA ER^−^) [49], colon cancer (COAD) [51], rectum cancer (READ) [51], bladder cancer (BLCA) [51], renal cell carcinoma (RCC) [51], cervical cancer (CESC) [51], uterine corpus endometrial cancer (UCEC) [51], malignant melanoma (MELA) [51], prostate cancer (PRAD) [60, 61], and ovarian cancer (OCAC) [62]. General information on these GWAS studies is provided in Additional File 2: Table S1.
Quality control for SNPs
All GWAS summary statistics were harmonized to the GRCh37 (hg19) reference genome prior to analysis. SNP-level quality control was performed following standard GWAS practices to ensure data consistency across traits. Filtering criteria are as follows: (i) exclusion of non-bipartite allele SNPs and SNPs with strand-ambiguous alleles; (ii) exclusion of SNPs without rs tags; (iii) deletion of duplicated SNPs, SNPs excluded in the 1000 Genomes Project, or with mismatched alleles; (iv) exclusion of SNPs within the major histocompatibility complex region at chr6: 28.5–33.5 Mb from LDSC analysis; (v) retention of SNPs with minor allele frequency (MAF) > 0.01.
Genome-wide genetic correlations analysis
Global genetic correlations (rg) between PSU phenotype and cancer were assessed by linkage disequilibrium score regression (LDSC) [63] and high-definition likelihood (HDL) [64]. LDSC quantifies the genome-wide covariance in SNP effect sizes between two traits, thereby capturing polygenic genetic correlation across the entire genome. The 1000 Genomes Project Phase 3 European super-population served as the linkage disequilibrium (LD) reference panel, and SNPs were filtered to the HapMap3 set recommended by the LDSC developers. The baselineLD_v2.2 model was applied to control for potential confounding due to functional annotation and LD structure. The LD scores used in LDSC were calculated from common SNPs (minor allele frequency > 0.01) in the 1000 Genomes reference panel. Standard errors were estimated using the block jackknife method, and the LDSC intercept was used to assess potential sample overlap or residual population stratification between datasets. All GWAS summary statistics were preprocessed with the “munge_sumstats.py” script to harmonize alleles, align effect directions, and remove ambiguous or mismatched SNPs. This procedure ensured that all datasets were aligned to a consistent genomic build (GRCh37/hg19) and reference allele orientation before downstream analyses. The reference panel including 1,029,876 quality-controlled HapMap3 SNPs was used for HDL analysis.
Identification of pleiotropic loci and genes
Pleiotropic analysis under composite null hypothesis (PLACO) analysis served to systematically identify genetic associations between PSU and cancer at SNP level. PLACO is a statistical approach specifically designed to detect pleiotropy, enabling the identification of shared genetic variants across multiple phenotypes [65]. The composite null hypothesis in PLACO assumes that a given SNP is associated with at most one trait, that is, either associated with trait 1 only, trait 2 only, or neither trait. The alternative hypothesis corresponds to a true bivariate association, where the SNP is simultaneously associated with both traits. PLACO tests this composite null using a product-based test statistic that combines Z-scores from the two GWASs, providing greater sensitivity for detecting shared associations while maintaining strict type I error control. We considered SNPs that reached genome-wide significance (P < 5 × 10^−8^) as pleiotropic variants, indicating that these SNPs show strong genetic associations across multiple phenotypes. Identification of pleiotropic variants is essential for uncovering the shared genetic basis of PSU and cancer. Functional Mapping and Annotation (FUMA) was used to map risk variants to specific genomic regions. FUMA localizes SNPs to specific genomic regions (i.e., risk loci), providing deeper insights into potential functions of these variants [66]. Lead SNPs were used to map nearby genes within each locus and generalized gene-set analysis of GWAS data served to identify biological functions of pleiotropic loci. Locus definition was conducted using the 1000 Genomes Project Phase 3 European (1000G Phase3 EUR) reference panel with the following parameter settings: maximum P-value for lead SNPs = 5 × 10⁻⁸, maximum P-value cutoff for candidate SNPs = 0.05, r^2^ threshold to define independent significant SNPs = 0.6, secondary r^2^ threshold to define lead SNPs = 0.1, minimum minor allele frequency (MAF) = 0, and maximum distance between LD blocks to merge into one locus = 250 kb.
Multi-marker analysis of genomic annotation (MAGMA) was conducted to determine pleiotropic genes via the implementation of linkage disequilibrium between markers and to detect multi-marker effects [67]. SNPs were mapped to genes according to the GENCODE v19 (GRCh37/hg19) annotation with a ± 50 kb window around gene boundaries, and the same 1000 Genomes Phase 3 European reference panel was used to account for linkage disequilibrium among markers. MAGMA first aggregates SNP-level associations into gene-level statistics and then evaluates enrichment across predefined biological pathways. 10,678 gene sets including curated gene sets (c2.all) and gene ontology (GO) terms (c5.bp, c5.cc, and c5.mf) from Molecular Signatures Database (MSigDB) were tested [68]. Bonferroni correction was used for all tested gene sets to avoid false positives (P < 0.05/10,678 = 4.68 × 10^−6^).
Colocalization analysis
R package coloc [69] served to determine co-localizations of association signals for PSU and cancer. For each of the shared SNPs between trait pairs, variants in the range of 500 Kb of the index SNP were extracted and the probability that the two traits share one common causal variant (H4) was calculated. Colocalization was considered for loci with a probability greater than 0.7. The posterior probability (PP) of multiple traits sharing the same SNP was estimated using a Bayesian divisive clustering algorithm implemented by the HyPrColoc package in R [70].
Tissue-enrichment analysis
Genome-wide tissue-specific enrichment analysis (GWTSEA) was conducted based on 30 general GTEx tissues [69] for MAGMA-derived genome-wide pleiotropic loci [68]. Considering that the brain was significantly enriched in all trait pairs, we further perform GWTSEA based on 13 GTEx brain tissues at the GTEx website (https://www.gtexportal.org/home/tissue/).
Single-cell level specificity
Single-cell disease relevance score (scDRS) relates scRNAseq data to polygenic disease risk at single-cell resolution, independently of the annotated cell type [71]. We applied scDRS (version v1.0.2) to assess cell-level disease associations (https://github.com/martinjzhang/scDRS). The scDRS framework estimates disease relevance at the single-cell level by integrating GWAS-based polygenic risk signals with single-cell transcriptomic data. It performs a gene set enrichment analysis using a gene set whose members are weighted by their association strength with the trait of interest, derived from an external method. In this study, the gene-level P-value output from MAGMA served as the input to the scdrs munge-gs function. For each cell, scDRS computes a raw disease relevance score based on the expression of trait-associated genes and then generates multiple Monte Carlo (MC) control scores from matched random gene sets with similar expression characteristics. A unified MC test is subsequently used to obtain an empirical P-value for each cell, identifying cells whose transcriptomic profiles show significant enrichment for disease-associated genes. The method further performs downstream analyses to identify (i) significant pre-annotated cell states based on aggregated group Z scores and (ii) individual genes whose expression levels correlate with the inferred disease relevance scores. All analyses were conducted using default parameter settings as recommended in the official repository.
Gene-based association analysis
We used MAGMA, transcriptome-wide association study (TWAS)-fusion, and summary-based Mendelian randomization (SMR) to identify common genes shared across trait pairs. Genome-wide gene-based association studies (GWGAS) were conducted using MAGMA [68], integrating SNP-based P-values from GWAS, including 19,427 protein-coding genes (NCBI 37.3 gene annotations). Gene association tests considered linkage disequilibrium (LD) of SNPs, ensuring accurate assessment of gene-level associations. Genes exhibiting a P-value below the threshold of 0.05 were selected for subsequent analysis.
TWAS for was performed using FUSION (http://gusevlab.org/projects/fusion/) [72]. Reference weights for gene expression in the target tissue were calculated with multiple prediction models in FUSION. SNPs within 1 Mb of a given gene were selected from GWAS summary statistics. The imputation Z score for the cis genetic effect on each trait was calculated using the formula ZTWAS = W’Z/(W’SW)1/2. Z represents GWAS summary Z-scores, W represents weights, and S the SNP-correlation covariance matrix. Genes with P-values < 0.05 after 5000 permutations were selected for further analysis.
SMR [73] associated GWAS summary-level data with expression quantitative trait loci (eQTL) studies to identify pleiotropic genes associated with complex traits. It employs SMR and heterogeneity in dependent instruments (HEIDI) methods to test pleiotropic associations between gene expression levels and complex traits of interest using summary-level data from GWAS and eQTL studies. This approach tests whether the magnitude of SNP effects on phenotypes is mediated by gene expression. Genes with a permutated P-value < 0.05 were selected for subsequent analysis.
Causal association analysis
Two-sample Mendelian randomization (MR) analysis was conducted to determine potential causal effects between PSU and cancer traits. Linkage disequilibrium (r^2^) clumping was employed in PLINK 1.9 to obtain independent significance SNPs (P < 5 × 10⁻⁸) for all exposure traits, using an r^2^ threshold of 0.001 within a 10,000 kb window [74] based on the 1000 Genomes Project Phase 3 reference panel. The strength of the instrumental variables (IVs) was evaluated using the proportion of variance explained (PVE) and the F statistic (F > 10) [75]. Causal effects between each trait pair were assessed using four MR methods: inverse variance weighted (IVW) [76], MR Egger [77], RAPS [78], and CAUSE [79]. Cochran’s Q statistics were applied to detect the effect size heterogeneity across the IVs [80]. A false discovery rate (FDR) of 5% was used as the threshold.
Mediation analysis
Two-step MR based mediation analysis was employed to disclose whether PSU mediates the causal pathway from hub gene to cancer outcome. The overall effect can be divided into an indirect effect (through mediators) and a direct effect (without mediators) effect [81]. The total effect of PSU on cancer was divided into 1) direct effects of PSU on cancer and 2) indirect effects mediated by PSU through the mediator. The proportion of mediating effect was obtained by dividing the indirect effect by the total effect, with 95% confidence intervals determined via the delta method.
Phenome-wide association study (PheWAS)
A PheWAS with the ExPheWas online tool (https://exphewas.statgen.org/) served to identify horizontal pleiotropy of potential drug targets and possible side effects [82]. Multiple corrections were performed, and a threshold of 5 × 10^−8^ was set as the default in the ExPheWas Portal to account for the potential for false positives.
Candidate drug prediction
Assessing protein-drug interactions was conducted using the Drug-Gene Interaction database (DGIdb, https://www.dgidb.org/) to screen potential drugs [83]. DGIdb is a database of drug-gene interactions that provides information on known or potential associations between genes and drugs. It is currently updated to version 5.0. DGIdb contains over 14,000 drug-gene interactions, involving 2600 genes and 6300 targeting drugs. Additionally, it includes 6,700 additional genes that are likely to become drug targets in the future.
Molecular docking analysis
Molecular docking was performed to evaluate the interactions between hub proteins and their corresponding candidate drugs predicted by the DGIdb database. Docking simulations were conducted using AutoDock Vina (version v1.1.2) [84], and protein structures were obtained from the Protein Data Bank (PDB) or AlphaFold when experimental structures were unavailable. Proteins were prepared by removing water molecules, adding polar hydrogens, and assigning Gasteiger charges using AutoDockTools. Ligand structures were retrieved from DGIdb databases and energy-minimized using the MMFF94 force field. A docking grid was defined to cover the predicted binding pocket of each protein. For each protein–drug pair, nine docking poses were generated, and the pose with the lowest binding energy was selected for further interpretation. Docking affinity scores (kcal/mol) and interaction profiles were analyzed to assess druggability, and top-ranked complexes were visualized using PyMOL (version v 3.1.3).
Software and packages
Statistical analysis was performed in R (version v4.1.1). LDSC analysis was conducted with “LDSC” software (version v1.0.1) [63]. PLACO was performed with the “PLACO” package [65]. Bayesian colocalization analysis was performed with the “coloc” package (version v5.2.1) [85]. Function analysis was performed with the FUMA web tool [66]. MAGMA gene and gene-set analysis were performed with the MAGMA software [67]. Two-sample MR analysis was conducted with “MendelianRandomization” (version v0.10.0) [86].
Results
Genetic correlations between PSU phenotypes and cancer
The present study interrogated shared genetic architectures between PSU and tumor entities in GWAS summary-level data. First, we evaluated the global genetic correlations between PSU and cancer trait pairs and identified shared pleiotropic loci. Furthermore, the causal relationship between PSU and cancer was detected by MR analyses. The shared biological mechanism of these trait pairs was then characterized by enriched pathways, relevant organs or tissues, and specific cell subtypes. Shared pleiotropic genes were subsequently obtained through TWAS and SMR analyses. Finally, we performed drug-target prediction based on the identified pleiotropic genes and constructed a regulatory network to highlight potential therapeutic targets (Fig. 1).Fig. 1. Schematic representation of the overall study design. Shared SNPs associated with psychoactive substance use (PSU) and pan-cancer entities were identified via LDSC and HDL. Pleiotropy of identified SNPs/loci was conducted with PLACO. Causal association between PSU and cancer was detected by MR analysis. Functional and expression analysis of SNPs/loci was addressed with MAGMA, data from the GTEx project, and scRNAseq datasets of cerebellum regions. Shared hub genes were analyzed for potential effects on tissue (PheWAS) and for the availability of clinical drugs
We first estimated the SNP-based heritability (h_2_^SNP^) for PSU and cancer traits. Phenotypes, sample size, ratio of case-to-controls, source, and PubMed ID are summarized in Additional file 2: Table S1. This cross-trait GWAS aggregated 6,145,925 samples with 598,931 cases, and case proportions of 0.49–56.43%. The results indicated significant heritability for all traits and reflected the polygenic character of each trait. We employed both LDSC and HDL to assess global genetic correlations (rg) for each trait pair composed of one PSU and one cancer subtype individually (Fig. 2A). A total of eight PSU traits including AlcUD, AspU, HypU, OpiU, CanUD, NicUD, CofU, and TeaU were cross-referenced with n = 20 tumor types, resulting in 34 shared trait pairs with significant genetic correlations identified with LDSC and HDL (Fig. 2B). PSU showed a positive genetic correlation with n = 11/20 cancer types. Notably, all PSU traits except CofU showed significant positive genetic correlations with LC-related traits. All BC traits exhibited remarkable negative genetic correlations with CofU (Additional file 2: Table S2 and Fig. 2B).Fig. 2. Global genetic correlations between psychoactive substance use and pan-cancer. A Genetic correlation analysis between PSU and cancer entities using LDSC and HDL. Correlation levels are indicated by the shape of the symbol, correlation strength by color-coding, and correlation directionality by left (negative) or right tilt (positive). B Shared trait pairs between LDSC and HDL are depicted in a Sankey diagram including PSU and the indicated cancer entities. Abbreviations: alcohol use dependence (AlcUD), nicotine use dependence (NicUD), cannabis use disorder (CanUD), opioid use (OpiU), aspirin use (AspU), hypnotics use (HypU), tea use (TeaU), and coffee use (CofU)
Shared loci and genes between PSU phenotypes and cancer
PLACO was employed to explore potential pleiotropic loci for each trait pair identified with LDSC and HDL. Subsequently, we conducted a functional mapping and annotation of genetic associations of PLACO results using FUMA [65] (Fig. 3A). A total of 97 pleiotropic genomic risk loci significantly correlated with PSU-cancer trait pairs were identified (P < 5 × 10^−8^) (Additional file 2: Table S3). We failed to identify significant risk loci in the two types of trait pairs “aspirin use-cancer” and “hypnotics use-cancer”. Several shared genomic loci were identified across multiple trait pairs—e.g., rs71658797 was shared between AlcUD-LC and AlcUD-LUSC as well as rs780092, which was shared between AlcUD-BRCA ER^−^ and AlcUD-OCAC (Additional file 2: Table S3). Colocalization analysis using single causal variant assumption (coloc analysis) [87] identified 22 of the 97 (22.68%) potential pleiotropic loci with posterior probability of the shared causal variant hypothesis H4 (PP.H4) values above 0.7, indicating colocalization (Additional file 2: Table S3).Fig. 3. Pleiotropic associations across alcohol use and pan-cancer. A Schematic representation of the study workflow designed to identify shared and causal loci/genes of trait pairs. B Circular network showing the pleiotropic landscape between alcohol use dependence (AlcUD) and estrogen receptor-negative breast cancer (BRCA-ER −), lung cancer (LC), lung squamous cell carcinoma (LUSC), and ovarian cancer (OCAC). The circular network includes SNP-affected chromosomal loci (yellow circles) and genes (blue circles) and distinguishes shared and causal loci (open/closed circles), and shared genes identified by positional mapping and by MAGMA analysis (open/closed circles)
It is noteworthy that some pleiotropic chromosomal regions were found to be shared between different trait pairs. For example, chromosomal regions 1p31.1, 2p23.3, and 15q15.3 were identified as shared genomic regions associated with multiple trait pairs. Specifically, 1p31.1 was linked to both AlcUD-LUSC and AlcUD-LC; 15q15.3 was shared by AlcUD-LUSC and AlcUD-LC; and 2p23.3 was associated with AlcUD-BRCA ER − and AlcUD-OCAC, indicating these regions play a crucial role in the genetic correlations between various cancer types and alcohol use traits (Additional file 2: Table S4).
To detect shared mechanisms of the identified loci, we mapped nearby genes according to the lead SNP in each locus based on its most significant trait association, and under the assumption of its linkage disequilibrium with other SNPs within the loci. Additionally, we performed a multi-marker effect analysis on the GWAS data using MAGMA [67] to explore shared pleiotropic genes across trait pairs. Summarizing the shared loci and genes, circular networks were generated to visualize the pleiotropic architecture of trait pairs (Fig. 3B and Additional file 1: Fig. S1). As exemplified for AlcUD, pairing with BRCA ER −, OCAC, LC, and LUSC was associated each with ≥ 3 loci and ≥ 17 genes (Fig. 3B). Enrichment results from MAGMA indicated that the pleiotropic loci are involved in nicotinic acetylcholine receptor, generation and development of neurons, cell cycle, and transcription regulation (Additional file 1: Fig. S2). Notably, the generation and development of neurons was strongly associated with all trait pairs and cell cycle as well as transcription regulation were detected in most of the trait pairs (Additional file 2: Table S5).
Tissue and cell-type specificity
Tissue-specific expression analysis using the GTEx database revealed that risk loci were enriched in brain tissue. We further explored 13 different brain region tissues and demonstrated that all trait pairs were significantly associated with the cerebellum (Additional file 2: Table S6). To detect the specificity of different trait pairs at the single-cell level, scDRS analysis was employed (Fig. 4A). A scRNAseq dataset of human brain cerebellum was collected and Purkinje cells, unipolar brush cells, neuroblasts, GABAergic neurons, macroglia, oligodendrocytes, granule cells, precursor granule cells, glutamatergic neurons, interneurons, microglial cells, and Bergmann glial cells were identified based on cell markers (Fig. 4B–C). The results revealed that scDRS of trait pairs were most strongly enriched in GABAergic neurons, granule cells, glutamatergic neurons, and interneurons, and were depleted in oligodendrocytes. These enriched cell subtypes constitute major excitatory and inhibitory circuits of the cerebellum and have been implicated in addictive behaviors, indicating that PSU and cancer trait pairs may share genetic risk through cerebellar neuronal pathways involved in synaptic transmission and behavioral regulation (Fig. 4D–E and Additional file 1: Fig. S3).Fig. 4. Single-cell specificity inferred from the shared signals between psychoactive substance use and pan-cancer. A Schematic representation of the study workflow. B UMAP of the cell annotation of scRNAseq dataset of brain cerebellum showing 12 cell types. C Dotplot showing specific markers of 12 brain cerebellum cell types. D UMAP representation of single-cell disease relevance z-scores (scDRS) of the indicated trait pairs. The color represents the disease score, where a darker color indicates a higher score, signifying greater disease enrichment for the specific cell cluster. E Heatmap representation of scDRS for 20 indicated trait pairs
Identification of actionable tissue-specific pleiotropic genes
TWAS and SMR analyses were conducted to interrogate gene-tissue effects for each PSU cancer trait pair. TWAS detects the association between genetically predicted mRNA expression and the GWAS trait, whereas SMR evaluates putative causal effects of gene expression on the trait using cis-eQTLs as instrumental variables under the MR method (Fig. 5A). A total of 109 genes identified by MAGMA were subsequently intersected with candidate genes from TWAS and SMR to screen hub genes between different PSU and cancer trait pairs. (Additional file 2: Table S7). We identified 21 pleiotropic genes across multiple trait pairs, with EFNA1 identified as a pleiotropic gene for six trait pairs and CHRNA2 in five trait pairs (Fig. 5B–G; Additional file 2: Table S8).Fig. 5. Shared gene signals between different PSU and cancer trait pairs. (A) Study design for identifying hub genes shared between different trait pairs. Transcriptome-wide association study (TWAS) and summary-based Mendelian randomization (SMR) analyses were first conducted separately for different PSU and cancer trait pairs using brain cerebellum tissue of GTEx database. The overlap between PSU- and cancer-associated genes in TWAS identified TWAS shared genes, while the overlap in SMR highlighted SMR shared genes with potential causal effects on both traits. Subsequently, the TWAS shared genes, SMR shared genes, and MAGMA shared genes (derived from previous MAGMA analyses of PSU and cancer trait pairs) were integrated to identify hub genes. These hub genes may represent key genetic components underlying the shared biological mechanisms between PSU and cancer. B–G Venn diagram representation of shared gene signals for alcohol use dependence (AlcUD; B), nicotine use dependence (NicUD; C), cannabis use disorder (CanUD; D), opioid use (OpiU; E), coffee use (CofU; F), and tea use (TeaU; G)
The causal association between PSU and cancer
MR analysis was applied to assess whether a causal relationship exists between PSU and cancer (Fig. 6A). The IVW-based MR analyses indicated a causal effect of PSU (AlcUD and NicUD) on cancer (OCAC and LC) risk (Fig. 6B–C; Additional file 2: Table S9). The risk of OCAC was observed to increase with higher genetic liability to AlcUD, with the causal effect assessed by the IVW method (OR = 1.216, 95% CI = 1.082–1.366, P = 0.001). Additionally, MR revealed a remarkable causal effect of NicUD on LC risk (IVW, OR = 1.124, 95% CI = 1.010–1.251, P = 0.032). Moreover, the scatter plot indicated a favorable concordance among different MR methods (IVW, MR Egger, RAPS, and CAUSE), suggesting a stable and reliable causal inference (Fig. 6D–E, Additional file 2: Table S10). To further explore whether the identified hub genes influence cancer risk through PSU, we employed mediation analysis based on the Two-step MR method. Based on the MR results, AlcUD-OCAC and NicUD-LC were selected for mediation analyses. According to the identified hub genes, HOXB2 was tested for the AlcUD-OCAC pair, while NSRP1, ZDHHC4, and EFCAB5 were tested for the NicUD-LC pair. It was observed that no significant indirect effects were detected across all tested genes (Additional file 2: Table S11), indicating no evidence supporting PSU as a mediator in the causal pathways from hub genes to OCAC or LC.Fig. 6. Causal association analysis. A Study design of Mendelian randomization (MR) analysis. B–C The forest plot shows causal associations between PSU and cancer by using MR analysis. D Scatter plot shows a significant causal relationship between AlcUD and OCAC risk. E Scatter plot shows a significant causal relationship between NicUD and LC risk
Drug target analysis
To screen potential drugs targeting 21 identified hub genes, we performed a PheWAS using ExPheWas at the gene level to assess whether hub genes have beneficial or deleterious effects on other traits. None of the three genes CHRNA2, HRH3, and PTK6 were significantly associated with other traits (P < 5E^−8^ for genomic association; Additional file 2: Table S12). This finding suggested that potential side effects of drugs acting on CHRNA2, HRH3, and PTK6 and the presence of horizontal pleiotropy in these genes are likely to be small. The DGIdb database was used to make predictions of potentially effective interventional drugs on CHRNA2, HRH3, and PTK6 (Fig. 7A and Additional file 2: Table S13).Fig. 7. Drug target analysis. A Shared genes HRH3, PTK6, and CHRNA2 and their interaction potential with the indicated approved and non-approved drugs (open/filed circles) are depicted. Circle size indicates the interaction potential from low to high.(B–D Molecular docking analysis of HRH3, CHRNA2, and PTK6 with their potential interacting compounds
To verify the prediction results from DGIdb, molecular docking analyses were performed for CHRNA2, HRH3, and PTK6. Docking simulations confirmed stable binding affinities between the predicted compounds and their respective proteins (Additional file 2: Table S14). The top five docking complexes for each of the hub proteins are shown in Fig. 7B–D. The largest number of potential drugs was identified for HRH3, among which GSK-189254 and PF-03654746 exhibited the strongest interactions, as supported by both DGIdb prediction and molecular docking analyses. In addition, PIPECURONIUM was identified as the most promising drug targeting CHRNA2 (Additional file 2: Table S14). Hence, genes identified as central hubs in the shared genetic architecture of PSU and cancers show traits of druggability that may be used in future pre-clinical targeting strategies.
Discussion
Alcohol and nicotine are estimated to affect up to 30% of individuals worldwide and to annually account for three and seven million deaths, respectively [88]. A substantial proportion of these deaths are related to the induction of malignancies such as lung cancer and HNSCC through carcinogenic compounds present in tobacco products and pro-inflammatory effects of alcohol [15, 16, 89]. However, far less is known about an innate, shared genetic architecture that may coordinately impact on PSU and cancer as (multi-loci) SNPs with mutual interactions and effect sizes [90–92]. Unlike carcinogen-induced mutations, an inherited genetic architecture enhances the likelihood of a trait through coding and non-coding SNP variants that affect gene expression [92, 93]. Underlying genetic architectures can promote comorbidities through pleiotropic genes that impact two or more phenotypes, epistasis and modifier genes influencing disease outcome depending on the genetic background, overlapping pathways, and genetic predispositions that affect individual behavior and responses to environmental cues.
Deeper understanding of genetic architectures provides relevant molecular insight into comorbidities, their interrelations, and may help uncover novel pharmaceutical targets and predictive markers [90, 92, 94, 95]. For example, the PANcreatic Disease ReseArch (PANDoRA) consortium identified 25 susceptibility loci for the occurrence of pancreatic cancer [93], which can be leveraged to understand their contribution to this devastating malignancy. Genetic susceptibility loci identified by GWAS were reported for radiation-induced oral mucositis [96], Barrett´s esophagus and esophageal adenocarcinoma [97], clear cell renal cell carcinoma [98], and ovarian cancer [99], among others. A genetic architecture of liver cirrhosis determined risk-associated loci that were implemented in a polygenic risk score to predict a transition from liver cirrhosis to hepatocellular carcinoma [100]. Potential therapeutic targets to treat alcohol-UD were identified through the integration of proteomic, transcriptomic, and GWAS data [94, 101]. Risk genes related to tobacco use and their association with medical outcomes as diverse as heart disease, pain, and viral infection (i.e., HIV) were described [102, 103]. Most of these findings are very recent and large-scale analysis of PSU and cancer linkage at the level of genetic architecture is yet unavailable to our knowledge.
In our genome-wide case–control meta-analysis, we describe for the first time a pleiotropy underlying the genetic architecture of PSU and cancer linkage. We report a total of 34 shared trait pairs genetically linking lung adeno- and squamous carcinoma, breast carcinoma (ER − and ER +), ovarian, cervical, and esophageal adenocarcinoma with the use of alcohol, nicotine, cannabis, opioids, aspirin, hypnotics, coffee, and tea. Cross-trait pleiotropy analysis using PLACO identified 13 significant risk loci shared across multiple PSU and cancer trait pairs. MAGMA analysis revealed shared biological mechanisms underlying PSU and cancer, including neuronal generation and development, cell cycle regulation, and transcriptional control. Further TWAS and SMR analyses identified 21 shared hub genes, which were subsequently incorporated into drug-target prediction, resulting in a regulatory network comprising three key druggable genes (HRH3, CHRAN2, and PTK6). Finally, MR analysis provided evidence for causal associations between AlcUD and OCAC, as well as between NicUD and LC.
Our analyses revealed remarkable positive global genetic correlations between alcohol dependence, nicotine dependence, and cannabis use disorder and lung cancer, consistent with findings from previous observational studies [104–107]. Further subtype analyses identified differences in genetic correlations across lung cancer subtypes (LUAD, LUSC, and SCLC), thereby extending prior research. Moreover, we observed a positive genetic correlation between cannabis use disorder and cervical cancer. As no observational studies to date have demonstrated a promoting effect of cannabis use disorder on cervical cancer, this novel finding warrants further investigation into the underlying biological mechanisms.
Based on PLACO pleiotropy analysis, we identified a series of genetic risk loci associated with both PSU and cancer. Among these, the 19q13.2 region was of particular interest, as previous studies have demonstrated that this locus may play a crucial role in both PSU and various cancers. For example, the 19q13.2 locus has been reported to be associated with nicotine dependence [108, 109] and has also been linked to increased risks of several cancers, including lung cancer [110–114].
Notably, among all PSU and cancer trait pairs, coffee consumption showed a remarkable negative genetic correlation with breast cancer, in line with previous reports suggesting that coffee intake may reduce the risk of breast cancer [36, 37]. Our finding determined 2p25.3, 15q26.1, and 16q12.2 as shared pleiotropic loci between coffee consumption and breast cancer. These loci have not been reported in previous studies and represent novel findings of the present research. Based on MAGMA analysis, we observed several shared functional pathways between coffee use and breast cancer. Coffee and its bioactive components, such as caffeine, have been shown to regulate multiple nuclear receptors, including the aryl hydrocarbon receptor (AHR) [115, 116], peroxisome proliferator-activated receptors (PPARs) [117], and estrogen receptor (ER) signaling [118]. These receptors are involved in caffeine metabolism and are also closely associated with the development and progression of breast cancer [119–122]. Therefore, genetic variations that affect coffee metabolism or response may influence breast cancer susceptibility by modulating nuclear receptor-mediated transcriptional networks.
Our MR analysis further supports a causal relationship between PSU and cancer risk. Specifically, results based on the IVW method showed that increased genetic liability to AlcUD was significantly associated with a higher risk of OCAC, while NicUD exhibited a significant causal effect on LC, which is consistent with previous epidemiological studies [105, 123]. The concordant results across multiple MR methods (IVW, MR-Egger, RAPS, and CAUSE) further strengthen the robustness of our causal inference. The causal links identified in this study highlight the importance of PSU as a modifiable behavioral risk factor in cancer prevention.
Pleiotropy is a central aspect of comorbidities, which we mapped to loci and SNPs acting on hub genes in GABAergic neurons, granule cells, glutamatergic neurons, and interneurons of the brain cerebellum. The cerebellum emerged as an important factor in cancers through cognitive, emotional, and systemic regulation of the immune system, response to (chronic) stress, inflammation, angiogenesis, shaping of the tumor microenvironment, and through reward mechanisms and addiction [124–132]. Immunosurveillance and inflammation are regulated through cerebellum-derived neural pathways including the hypothalamic–pituitary–adrenal (HPA) pathway. These regulatory mechanisms are usurped by tumors for purposes of immune evasion [125, 133], leading to enhanced cortisol and stress hormones that have immunosuppressive functions supporting tumor outgrowth [134, 135]. The cerebellum further regulates a higher-order cognitive and emotional circuitry that promotes depression and anxiety when dysfunctional. Thereby, it negatively impacts on the patients’ outcome, compliance to treatment, and predisposition to addiction [136–138]. Based on all these implications in cancer outcome, approaches to re-install functional cerebellar activities are under development, including transcranial magnetic stimulation (TMS), cerebellar neurofeedback, and cognitive rehabilitation to revert dysfunctions [127].
In the present study, numerous approved and non-approved drugs were identified by using the GDIdb database for hub genes CHRNA2, HRH3, and PTK6 that are pleiotropic genes affecting several PSU-cancer trait pairs. CHRNA2 encodes the α2 subunit of neuronal nicotinic acetylcholine receptors (nAChR), which are ion channels activated upon binding of acetylcholine or nicotine. Knockout of CHRNA2 in female mice resulted in an increased cued fear conditioning following acute nicotine administration [139], whereas hypersensitive CHRNA2 hampered contextual fear conditioning [140]. CHRNA2 is affected by a lead SNP determined in a large-scale GWAS of cannabis UD and a missense mutation from Threonine 22 to Isoleucine is associated with addiction to nicotine [48, 141, 142]. Enhanced expression of CHRNA2 in prostate cancer correlated with poor response to immune checkpoint inhibition [143], and SNPs in the CHRNA2 locus predict susceptibility to lung cancer [50]. HRH3 is up-regulated in the pre-frontal cortex during social isolation-induced chronic stress [144], in lung cancer stem cells [145], and can be targeted with numerous drugs, according to the GDIdb and to reports of its inhibition in conjunction with alcohol UD [146]. In line with the effects of CHRNA2, HRH3 was shown to exert a negative regulation on peripheral immune cells following inflammatory signals [147]. Enhanced PTK6 expression correlates with poor outcomes and/or drives oncogenic progression of renal cell, hepatocellular, breast, ovarian, and prostate carcinoma [148–153]. Accordingly, inhibitory targeting of PTK6 has been interrogated in multiple cancers [154]. Furthermore, PTK6 has been recently associated with pathological drinking behavior and schizophrenia, major depression, and bipolar disorder [155].
Thus, our findings suggest an implication of the PSU-cancer genetic architecture in influencing behavioral and non-behavioral aspects with repercussions on substance use and cancer [156–158]. Inhibition of hub genes with pleiotropic effects in PSU-cancer trait pairs represents a potential avenue for future research into preventing comorbidities entailed by trait linkage. This is particularly of interest with respect to cannabinoids and opioids, as they are in use for the treatment of cancer patients to enhance appetite and reduce disease-entailed pain. Furthermore, defined SNPs and hub gene expressions may be investigated as potential markers to identify patients suffering from psychoactive drug abuse or from cancer who are at increased risk to develop respectively linked illnesses. Such stratification could be explored as a framework for improved personalized medicine and early detection to improve the prognosis of patients.
Limitations
The genetic architecture of illnesses and their linkage depend on genetic ancestry, which is not covered in an unbiased manner in current GWAS, as studies include mostly individuals of European ancestry [92, 159, 160]. Hence, future studies are needed in non-European populations. Further, a medical benefit of targeting hub genes associated with PSU-cancer trait pairs remains to be validated in experimental and clinical settings, which represents a challenging task.
Conclusions
This large post-GWAS study identifies a shared genetic architecture between psychoactive substance use (PSU) and cancer risk. The findings highlight genes linked to the brain’s reward system and social behavior, primarily expressed in the cerebellum, including three druggable targets. These results offer new insights into an underexplored genetic basis that predisposes individuals to both PSU and cancer, potentially paving the way for combinatorial treatment approaches for these common comorbidities.
Supplementary Information
Additional file 1. Figures S1–S3.Additional file 2. Tables S1–S14. Table S1. Data sources. Table S2. Genetic correlation analysis conducted by LDSC and HDL. Table S3. Shared pleiotropic loci identified by PLACO. Table S4. Shared pleiotropic loci among different trait-pairs. Table S5. MAGMA Gene-set analysis. Table S6. MAGMA tissue-specific analysis. Table S7. Tissue-specific pleiotropic genes. Table S8. Hub pleiotropic genes across multiple trait-pairs. Table S9. Causal association analysis by MR method. Table S10. Summary of CAUSE results. Table S11. Two-step Mendelian randomization mediation analysis. Table S12. Phenome-Wide Association Study. Table S13. Drug target analysis by DGIdb database. Table S14. Molecular docking analysis.Additional file 3. STROBE-MR-checklist.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gharahkhani P, Fitzgerald RC, Vaughan TL, Palles C, Gockel I, Tomlinson I, et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett's oesophagus: a large-scale meta-analysis. https://www.ebi.ac.uk/gwas/studies/GCST 003739. (2016). 10.1016/S 1470-2045(16)30240-6PMC 505245827527254 · doi ↗ · pubmed ↗
- 2Schumacher FR, Al Olama AA, Berndt SI, Benlloch S, Ahmed M, Saunders EJ, et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. https://www.ebi.ac.uk/gwas/studies/GCST 00608. (2018).10.1038/s 41588-018-0142-8PMC 656801229892016 · doi ↗ · pubmed ↗
- 3Cannon M, Stevenson J, Stahl K, Basu R, Coffman A, Kiwala S, et al. DG Idb 5.0: rebuilding the drug-gene interaction database for precision medicine and drug discovery platforms. Nucleic Acids Res. 2024;52(D 1):D 1227-D 35.10.1093/nar/gkad 1040 PMC 1076798237953380 · doi ↗ · pubmed ↗
- 4Dong J, Maj C, Tsavachidis S, Ostrom QT, Gharahkhani P, Anderson LA, et al. Sex-Specific Genetic Associations for Barrett's Esophagus and Esophageal Adenocarcinoma. Gastroenterology. 2020;159(6):2065–76 e 1.10.1053/j.gastro.2020.08.052PMC 905745632918910 · doi ↗ · pubmed ↗
- 5Cuellar-Partida G, Lu Y, Dixon SC, Australian Ovarian Cancer S, Fasching PA, Hein A, et al. Assessing the genetic architecture of epithelial ovarian cancer histological subtypes. Hum Genet. 2016;135(7):741–56.10.1007/s 00439-016-1663-9PMC 497607927075448 · doi ↗ · pubmed ↗
- 6Yin J, Rockenbauer E, Hedayati M, Jacobsen NR, Vogel U, Grossman L, et al. Multiple single nucleotide polymorphisms on human chromosome 19q 13.2–3 associate with risk of Basal cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2002;11(11):1449–53.12433725 · pubmed ↗
- 7Berg M, Agesen TH, Thiis-Evensen E, group IN-s, Merok MA, Teixeira MR, et al. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol Cancer. 2010;9:100.10.1186/1476-4598-9-100PMC 288534320459617 · doi ↗ · pubmed ↗