HMPA-Enabled Direct γ′-Arylation of Cyclic Vinylogous Esters
Yan-Xun Li, Wei-Ting Zhao, Yen-Ku Wu

TL;DR
A new palladium-catalyzed method enables selective γ′-arylation of cyclic vinylogous esters using HMPA as a regioselectivity additive.
Contribution
A Pd-catalyzed γ′-arylation method with HMPA for regioselective modification of cyclic vinylogous esters is introduced.
Findings
The method works with a wide range of (hetero)aryl bromides and CVE substrates.
γ′-aryl CVEs can be converted to α-arylcycloalkenones via Stork–Danheiser transposition.
Pd(dba)2/cataCXium A and HMPA enable regioselective γ′-arylation.
Abstract
Controlling regioselectivity in deprotonative arylation of conjugated carbonyl systems remains a longstanding challenge. We report a Pd-catalyzed γ′-arylation of cyclic vinylogous esters (CVEs), enabled by Pd(dba)2/cataCXium A and hexamethylphosphoramide (HMPA) as a regioselectivity-governing additive. The method accommodates a broad range of (hetero)aryl bromides and CVE substrates. The resulting γ′-aryl CVEs were further converted to α-arylcycloalkenones via Stork–Danheiser transposition.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
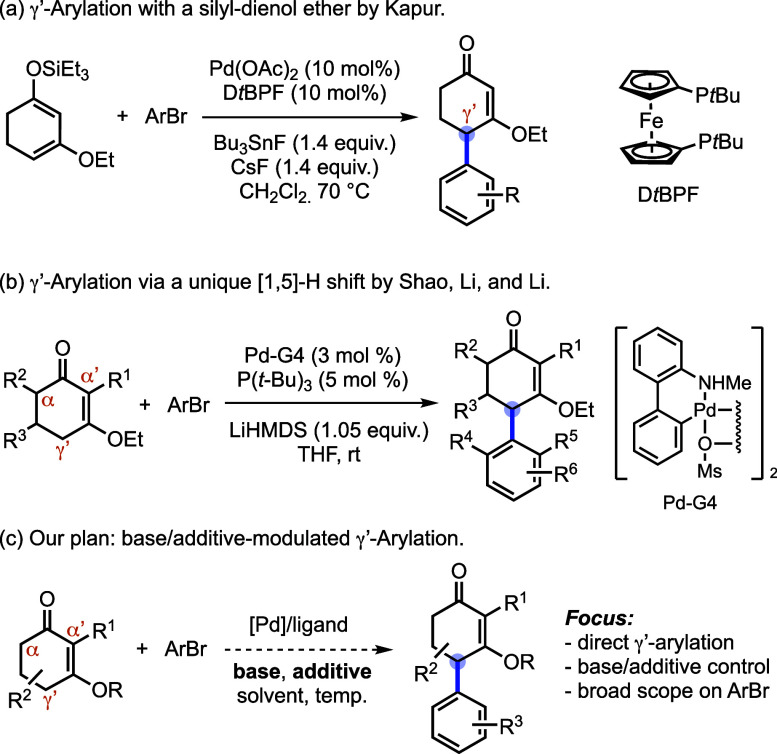 Figure 1
Figure 1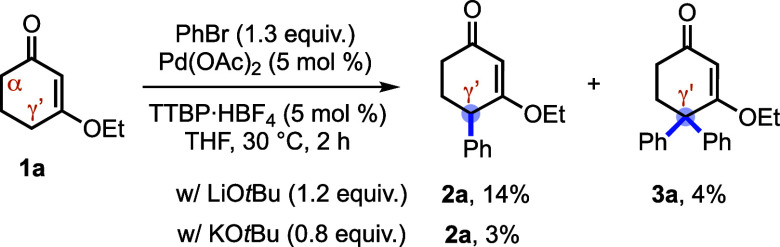 Figure 2
Figure 2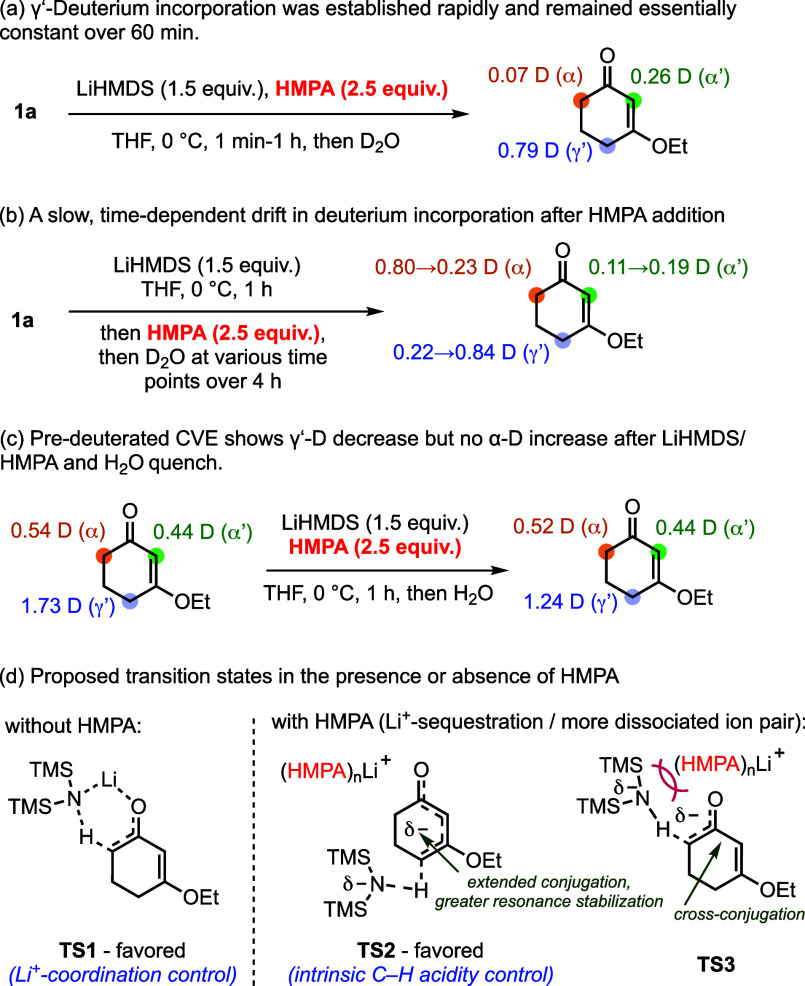 Figure 3
Figure 3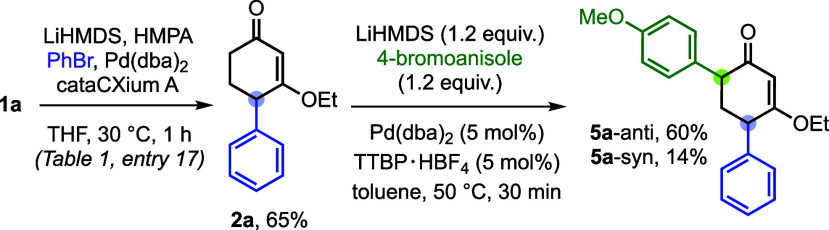 Figure 4
Figure 4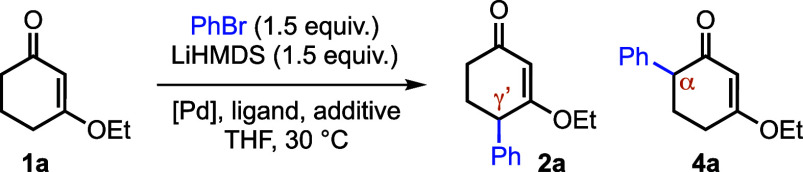 Figure 5
Figure 5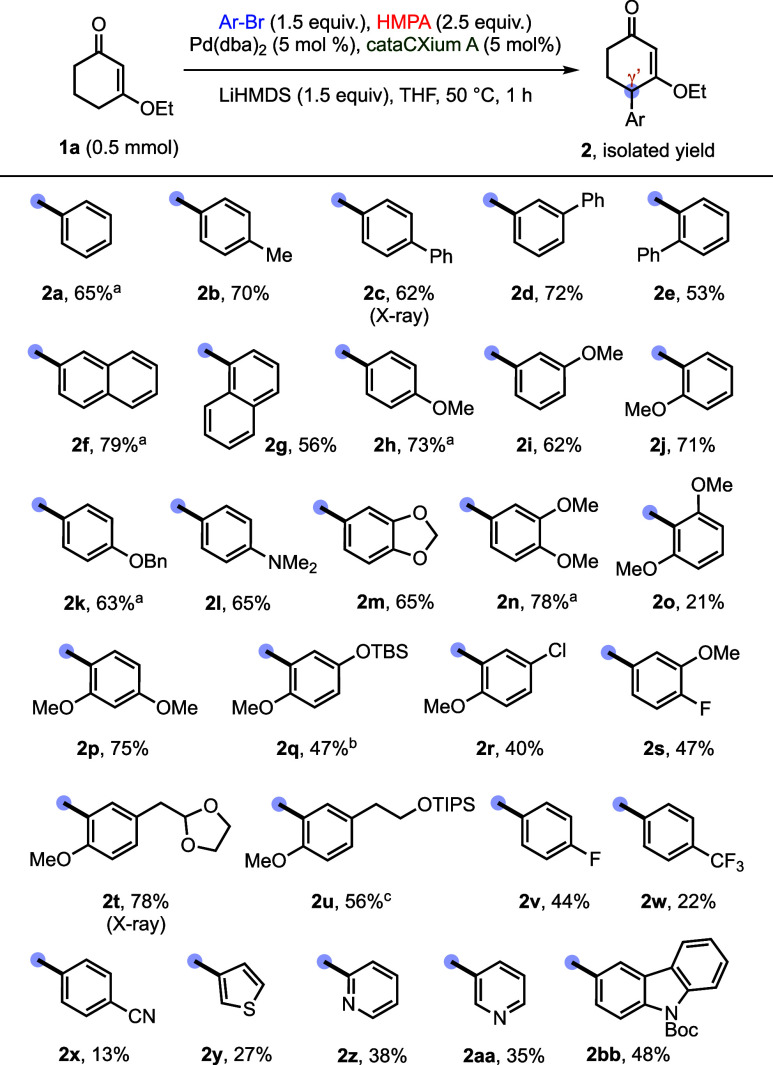 Figure 6
Figure 6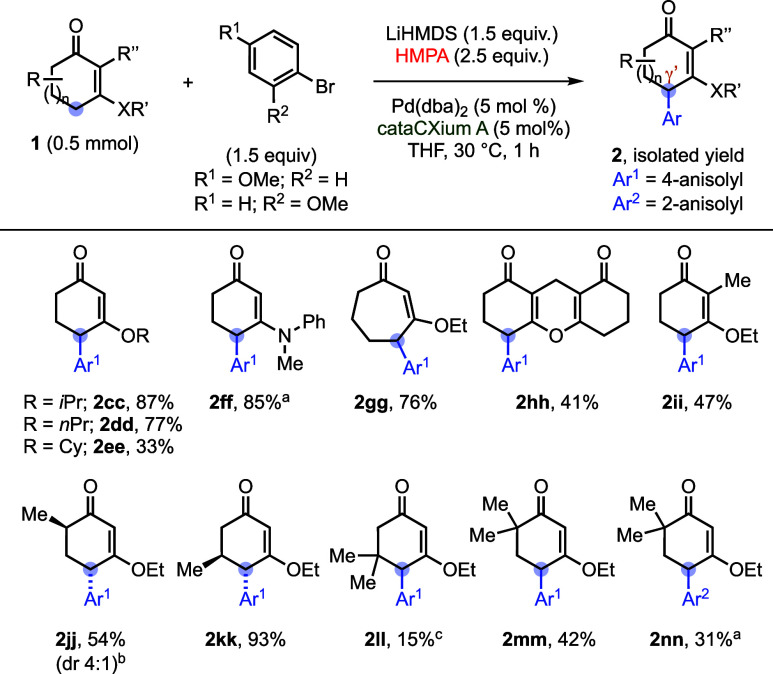 Figure 7
Figure 7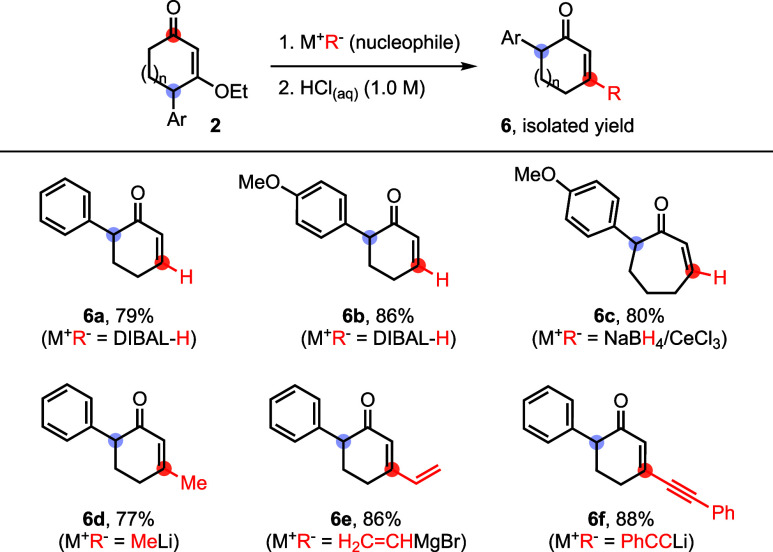 Figure 8
Figure 8 Figure 9
Figure 9 Figure 10
Figure 10- —National Science and Technology Council10.13039/501100020950
- —National Science and Technology Council10.13039/501100020950
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic Cross-Coupling Reactions · Catalytic C–H Functionalization Methods · Catalytic Alkyne Reactions
Aryl groups are ubiquitous in functional organic materials and small-molecule drugs, driving sustained interest in arylation methods.? While transition-metal catalysis has made aryl–carbon bond formation routine in many settings,? selective arylation at aliphatic carbon centers remains particularly valuable because it converts readily available carbonyl precursors into three-dimensional aryl-containing architectures. Among these transformations, Pd-catalyzed α-arylation of deprotonated carbonyl derivatives with aryl (pseudo)halides has become a benchmark strategy for stitching aromatic and alkyl fragments together.?
In contrast, catalytic deprotonative α- or γ′-arylation of conjugated carbonyl systems remains comparatively underdeveloped, largely because regioselectivity is difficult to control and competitive conjugate addition can erode efficiency.? Nevertheless, selective arylation at these positions would furnish versatile products that retain both an alkenyl handle and a carbonyl group for downstream diversification.? Achieving reliable regiocontrol in the arylation of conjugated carbonyl compounds would therefore open access to underexplored, three-dimensional arylated scaffolds.
Cyclic vinylogous esters (CVEs) represent a distinctive class of conjugated carbonyl systems that feature both α- and γ′-protons amenable to deprotonation, offering multiple opportunities for site-selective functionalization.? In the context of deprotonative arylation, Zhang,? Lautens,? and our laboratories? have reported Pd-catalyzed α-arylation of CVEs with haloarenes. We subsequently showed that Pd(OAc)2/tris(1-adamantyl)phosphine enables polyarylation of CVEs; when α’-alkyl-substituted CVEs were used, γ′-reactivity could be accessed to furnish α,γ′-diarylated and α,α,γ′-triarylated products under appropriate conditions.? Importantly, however, these cascade processes proceed through initial α-arylation and require α’-alkyl substitution to unlock γ′-functionalization. Kapur and co-workers addressed γ′-arylation via a two-step sequence in which γ′-selectivity is preset through formation of conjugated silyl dienol ethers prior to a modified Kuwajima–Urabe arylation (Schemea).? More recently, Shao et al. reported a catalytic γ′-arylation of CVEs in which regioselectivity is proposed to arise from a [1,5]-H shift of organopalladium intermediates; this platform is most effective for α-unblocked CVEs and ortho-substituted aryl bromides (Schemeb).? Overall, these studies underscore the continuing need for a direct, condition-controlled γ′-arylation of CVEs that is broadly applicable to CVE substrates and bromoarenes, including α,α-disubstituted CVEs and non-ortho aryl bromides (Schemec). In addition, we envisioned that γ′-aryl CVEs could be leveraged through Stork–Danheiser transposition? to furnish α-arylcycloalkenones, a motif prevalent in natural products and bioactive small molecules.
We selected 3-ethoxy-2-cyclohexenone (1a) and bromobenzene as model substrates to evaluate conditions for Pd-catalyzed γ′-arylation of CVEs. Initial screening focused on bases capable of generating the requisite γ′-dienolate. Under our standard catalytic manifold,? metal alkoxides furnished γ′-arylated product 2a only in low yield (Scheme), and in one case a γ′,γ′-diarylated byproduct 3a was also observed. Although these results suggested that γ′-selective arylation was feasible, further optimization of alkoxide-based conditions proved unproductive, prompting us to examine alternative base/additive combinations.
Guided by our prior Pd-catalyzed α-arylation of CVEs using lithium amide bases, we evaluated LiHMDS in combination with polar additives to modulate regioselectivity. The additive proved decisive: DMPU afforded γ′-arylated product 2a in 28% yield but also generated the competing α-arylation product 4a in 13% yield (Table, entry 1), whereas 12-crown-4 largely suppressed productive coupling (entry 2). In contrast, HMPA markedly improved both efficiency and γ′/α selectivity, delivering 2a in 45% yield with only 5% of 4a at 2.5 equiv (entry 3). Deviating from this HMPA loading (1.5 or 3.5 equiv) diminished the outcome (entries 4 and 5). With HMPA identified as the optimal additive, we next surveyed Pd sources and ligands. Pd(dba)2 was uniquely effective among the catalysts examined: Pd(OAc)2 was ineffective (entry 6), while common precatalysts provided only modest yields (entries 7–9). Ligand effects were pronounced; BINAP and P(oTol)3 gave only trace conversion (entries 10 and 11), whereas bulky dialkylbiaryl phosphines improved the reaction. Both XPhos and PAd_2_ nBu (cataCXium A)? furnished 2a in 54% yield, but PAd_2_ nBu further suppressed formation of the α-arylation product 4a (8% vs 2%; entries 13 and 14). Lowering the catalyst/ligand loading reduced efficiency (entries 15–16). Finally, premixing 1a with HMPA and LiHMDS to generate the dienolate prior to catalyst addition increased the yield of 2a to 65% (entry 17); the mass balance is accounted for by intractable byproducts. Under otherwise identical conditions, chlorobenzene was substantially less reactive than bromobenzene (entry 18), highlighting that the current system is optimized for aryl bromides. Control experiments confirmed that both Pd(dba)2 and PAd_2_ nBu are required for C–C bond formation.
The scope of aryl bromides was evaluated with 1a. Using entry 17 (Table) as the optimized baseline, we found that a modest temperature increase improved conversion for several substrates. Therefore, the scope studies were conducted at 50 °C unless otherwise noted. Electron-neutral aryl bromides generally performed well, furnishing γ′-arylated products in 53–79% yield (2a–g). Electron-rich aryl bromides were similarly effective, typically providing 2h–n and 2p–u in 40–78% yield. Steric effects were well tolerated for mono-ortho-substituted aryl bromides (2e, 2g, 2j, 2p, 2q, 2r, 2t, and 2u); however, the doubly ortho-substituted 2,6-dimethoxy aryl bromide led to a pronounced drop in yield (2o, 21%). Notably, the reaction displayed useful chemoselectivity: despite the increased steric congestion at the C–Br bond (ortho to the methoxy group), 2-bromo-4-chloroanisole underwent coupling exclusively at bromine, leaving the more accessible aryl chloride intact for further functionalization and furnishing 2r in 40% yield. In contrast, aryl bromides bearing strongly electron-withdrawing substituents were less competent: para-F, para-CF_3_, and para-CN substrates delivered products 2v–x in 13–44% yield; crude NMR analysis suggested appreciable aromatization of the CVE scaffold, producing 3-alkoxyphenol byproducts from direct CVE dehydrogenation and via γ′-arylation followed by aromatization. Additional variations in Pd source, ligand, or temperature did not substantially improve this subset. The protocol also enabled γ′-heteroarylation to afford products 2y–2bb in moderate yields. Finally, the reaction was readily scalable, delivering 2q (3.6 mmol) and 2u (1.0 mmol) in synthetically useful yields.
We next evaluated the scope of cyclic vinylogous esters (CVEs) using 4-bromoanisole and 2-bromoanisole as representative aryl donors (Table). Variation of the alkoxy substituent on the CVE was tolerated: i-propyloxy and n-propyloxy derivatives afforded 2cc and 2dd in good yields, whereas the more bulky cyclohexyloxy analogue gave diminished efficiency (2ee, 33%) and was accompanied by substantial decomposition/intractable material, consistent with increased steric congestion proximal to the γ′-position. Notably, a cyclic vinylogous amide also proved compatible, delivering 2ff in 85% yield. The influence of ring size was then examined. A seven-membered CVE underwent γ′-arylation smoothly to provide 2gg in 76% yield, while a 3-ethoxy-2-cyclopentenone substrate resulted in a complex mixture under the standard conditions. The protocol was further applicable to 1,8-dioxo-octahydroxanthene, furnishing the monoarylation product 2hh in moderate yield. We further probed substitution effects on the CVE scaffold. An α′-methyl-substituted substrate furnished 2ii, and an α-methyl CVE gave 2jj as a pair of diastereomers (dr = 4:1). In contrast, a β-methyl-substituted CVE provided 2kk with higher diastereoselectivity, plausibly reflecting a closer steric influence of the stereocontrol element. As illustrated by 2ll, a neighboring all-carbon quaternary center reduced the efficiency of γ′-arylation. Importantly, γ′-arylation of α,α-disubstituted CVEs was achieved to afford 2 mm (42%) and 2nn (31%). This substrate class (lacking an α-H) notably differs from the scope reported by Shao et al., in which α,α-disubstituted CVEs were not compatible (Schemeb).? Therefore, these results are consistent with a mechanism in which regioselectivity is established at the deprotonation stage to form a γ′-dienolate that subsequently undergoes Pd-catalyzed γ′-arylation.
To probe whether regioselectivity is established at the deprotonation stage, we performed D_2_O-quenching experiments on 1a in the absence of Pd catalyst and bromoarene. When a mixture of 1a, LiHMDS, and HMPA was quenched with D_2_O at different time points, deuterium incorporation occurred predominantly at the γ′-position, with ∼80% γ′-deuterium incorporation maintained across 60 min. (Schemea). This time-independent deuteration profile is consistent with direct and rapid γ′-selective deprotonation under HMPA-containing conditions.? In contrast, in the absence of HMPA, an analogous experiment afforded primarily α-deuterated 1a, in line with the prevailing view that lithium amides preferentially deprotonate CVEs at the α-position via a six-membered, coordination-assisted transition structure (TS1) to generate the kinetic dienolate.? In a complementary experiment, 1a was first reacted with LiHMDS in the absence of HMPA for 1 h, followed by addition of HMPA and subsequent D_2_O quenching at various time points (Schemeb). Under these conditions, the initial α-enriched deuteration pattern gradually evolved, and γ′-deuterium incorporation increased steadily, becoming dominant only after prolonged aging (>150 min; see Supporting Information). This time-dependent drift indicates that, once HMPA is introduced after initial deprotonation, interconversion among dienolate manifolds becomes feasible but proceeds on a comparatively slow time scale. Importantly, these results contrast sharply with the experiment in which HMPA is present from the outset, where the γ′-deuteration level is established rapidly and remains essentially unchanged.
To further assess possible H/D scrambling under the deprotonation conditions, a predeuterated CVE (γ′ = 1.73 D, α = 0.54 D) was subjected to LiHMDS/HMPA and then quenched with H_2_O (Schemec). The recovered material retained essentially the same α-deuterium content (0.54 to 0.52 D), while the γ′-deuterium content decreased (1.73 to 1.24 D), consistent with γ′-selective deprotonation/reprotonation. Notably, no measurable increase of deuterium at the α position was observed, arguing against significant intramolecular 1,5-H(D) transfer under these conditions and supporting deprotonation-controlled regioselectivity in our system. Taken together, these results suggest that HMPA governs the site of deprotonation and thereby controls the γ′/α selectivity observed in the catalytic arylation.? We propose that HMPA solvates Li^+^ to promote a more dissociated ion pair,? disrupting the pathway leading to TS1. Under such conditions, deprotonation at the γ′-position (TS2) becomes preferred to α-deprotonation (TS3), as the transition structure leading to an extended conjugated dienolate is better stabilized than that leading to a cross-conjugated dienolate.? Thus, when Li^+^ sequestration overrides carbonyl coordination, deprotonation proceeds primarily under the substrate’s intrinsic C–H acidity control, shifting the kinetic deprotonation manifold from TS1 to γ′-deprotonation via TS2.
As shown in Scheme, the present γ′-arylation can be readily combined with an established α-arylation protocol, ?,? enabling regioselective installation of two different aryl groups on the CVE core. This two-step sequence furnished the diarylated products 5a (anti/syn) in good yield (dr = 4:1). Notably, the order of operations is critical: the sequence must begin with γ′-arylation and be followed by α-arylation, because preliminary experiments showed that the α-monoarylated CVE is not competent under the γ′-arylation manifold. By comparison, our previously reported cascade polyarylation is restricted to α′-alkyl-substituted CVEs and provides reduced control over regioselectivity in the second arylation step.? These results further highlight that the present protocol enables complete and programmable control over site selectivity in deprotonative arylation of CVEs.
To further expand synthetic utility, we explored Stork–Danheiser transposition of γ′-arylated CVEs as an entry to α-aryl cycloalkenones. This strategy is synthetically attractive because direct α-arylation of cycloalkenones remains challenging and underdeveloped. In this sequence, carbon and hydride nucleophiles, such as MeLi, a Grignard reagent, PhCCLi, NaBH_4_, and DIBAL-H, added to the CVE carbonyl, and the resulting intermediates underwent acid-mediated hydrolysis to furnish the corresponding α-aryl cycloalkenones (Table). Overall, the combined γ′-arylation/Stork–Danheiser transposition provides a modular route to versatile carbocyclic building blocks 6a–6f.
In summary, we have developed a condition-controlled, γ′-selective monoarylation of cyclic vinylogous esters. The method can be sequenced with α-arylation to enable programmable installation of distinct aryl groups on the CVE scaffold, and it also provides access to α-aryl cycloalkenones via Stork–Danheiser transposition. Ongoing studies are focused on elucidating the role of HMPA and related polar additives in remote deprotonative arylation of conjugated carbonyl systems.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Aldeghi M.Malhotra S.Selwood D. L.Chan A. W.Two- and three-dimensional rings in drugs Chem. Biol. Drug Des.20148345046110.1111/cbdd.1226024472495 PMC 4233953 · doi ↗ · pubmed ↗
- 2a Hassan A.Sévignon M.Gozzi C.Schulz E.Lemaire M.Aryl–Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction Chem. Rev.20021021359147010.1021/cr 000664 r 11996540 · doi ↗ · pubmed ↗
- 3a Sivanandan S. T.Shaji A.Ibnusaud I.Seechurn C. C. C. J.Colacot T. J.Palladium-Catalyzed α-Arylation Reactions in Total Synthesis Eur. J. Org. Chem.20152015384910.1002/ejoc.201403301 · doi ↗
- 4a Saini G.Kapur M.Palladium-catalyzed functionalizations of acidic and non-acidic C(sp 3)–H bonds – recent advances Chem. Commun.2021571693171410.1039/D 0CC 06892 F 33492315 · doi ↗ · pubmed ↗
- 5a Hyde A. M.Buchwald S. L.Palladium-Catalyzed γ-Arylation of β,γ-Unsaturated Ketones: Application to a One-Pot Synthesis of Tricyclic Indolines Angew. Chem., Int. Ed.20084717718010.1002/anie.20070452918033712 · doi ↗ · pubmed ↗
- 6a Huang G.Kouklovsky C.de la Torre A.Gram-Scale Enantioselective Synthesis of (+)-Lucidumone J. Am. Chem. Soc.2022144178031780710.1021/jacs.2c 0876036150082 · doi ↗ · pubmed ↗
- 7Zhao Y.Zhou Y.Liang L.Yang X.Du F.Li L.Zhang H.Palladium-Catalyzed Sequential Arylation and Allylic Alkylation of Highly Functionalized Ketones: A Concise Synthesis of Mesembrine Org. Lett.20091155555810.1021/ol 802608 r 19117400 · doi ↗ · pubmed ↗
- 8Johnson T.Pultar F.Menke F.Lautens M.Palladium-Catalyzed α-Arylation of Vinylogous Esters for the Synthesis of γ,γ-Disubstituted Cyclohexenones Org. Lett.2016186488649110.1021/acs.orglett.6b 0339427978660 · doi ↗ · pubmed ↗