Base-Promoted Conversion of Propargylic Alcohols to 1,3-Enynes
Morgane Delattre, Milena Wiegand, Qian Wang, Jieping Zhu

TL;DR
A new base-promoted method converts propargylic alcohols into 1,3-enynes with high efficiency and compatibility with various functional groups.
Contribution
A novel base-mediated conversion of TMS ethers of propargylic alcohols to 1,3-enynes is introduced.
Findings
Base-promoted conversion of TMS ethers of propargylic alcohols to 1,3-enynes occurs in high yields.
The method is compatible with a broad range of functional groups.
Lithium acetylide trapping with electrophiles provides access to functionalized internal 1,3-enynes.
Abstract
Under acidic conditions, propargylic alcohols undergo Meyer–Schuster or Rupe rearrangements to afford two isomeric α,β-unsaturated ketones. Herein, we disclose a mechanistically distinct base-mediated regioselective conversion of TMS ethers of propargylic alcohols to 1,3-enynes in high yields with broad functional-group compatibility. Alternatively, trapping of the in situ-generated lithium acetylide with electrophiles enables access to functionalized internal 1,3-enynes. Owing to the ready accessibility of propargylic alcohols, this method provides a practical and attractive entry to synthetically valuable 1,3-enynes.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
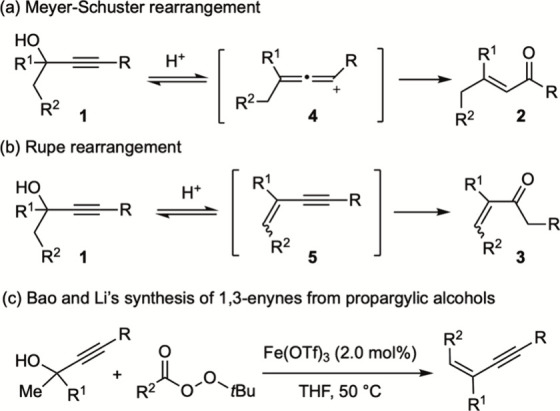 Figure 1
Figure 1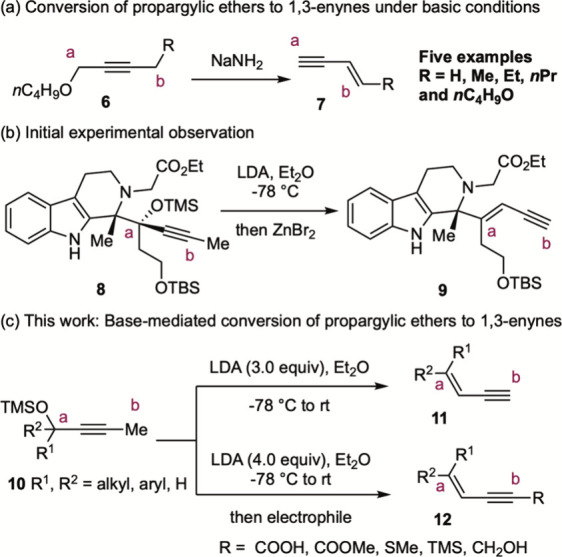 Figure 2
Figure 2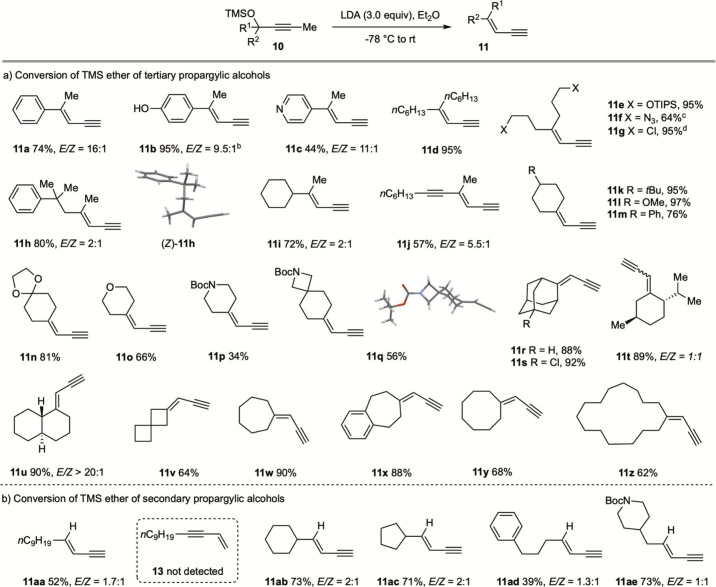 Figure 3
Figure 3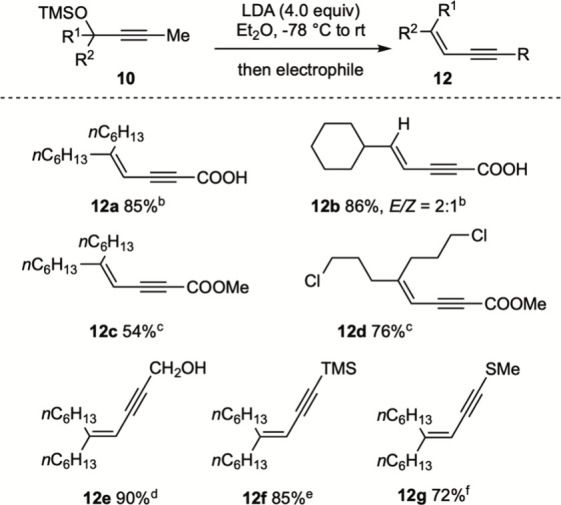 Figure 4
Figure 4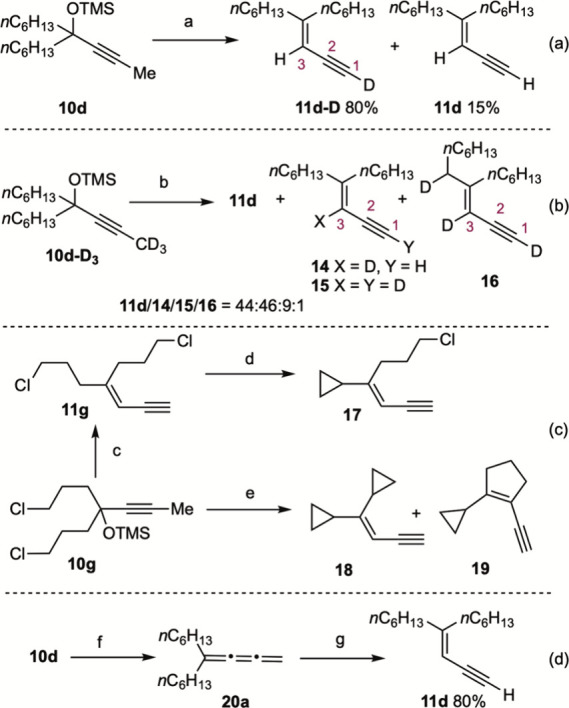 Figure 5
Figure 5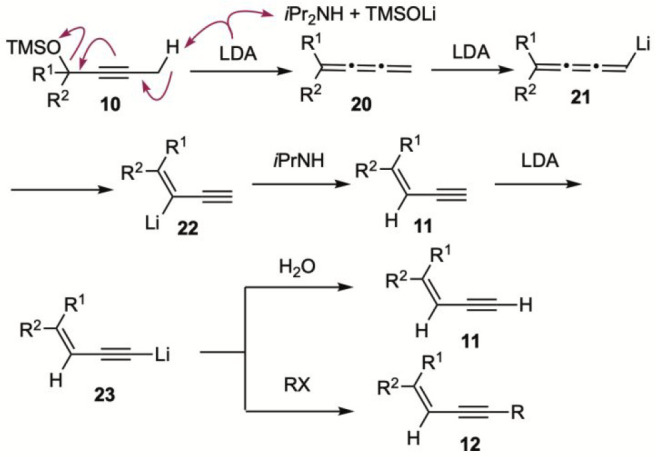 Figure 6
Figure 6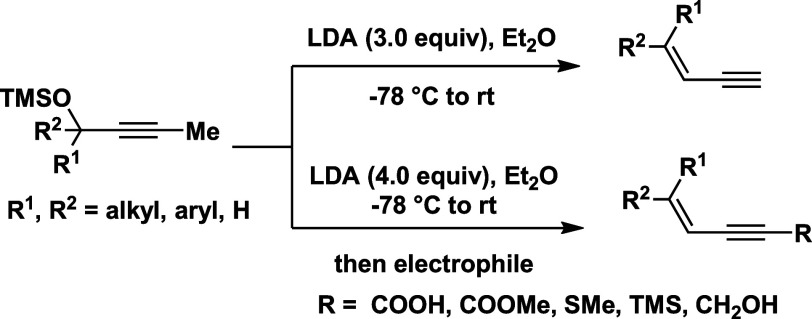 Figure 7
Figure 7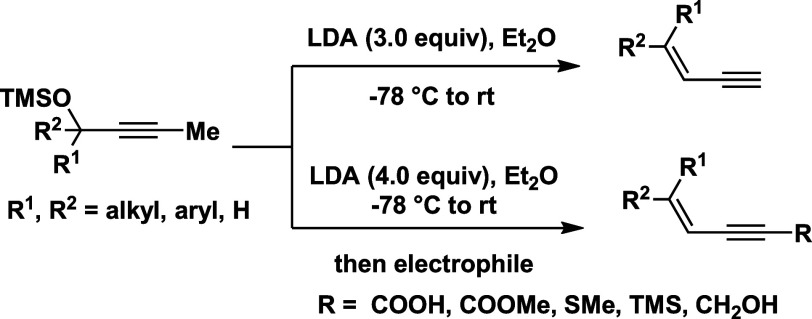 Figure 8
Figure 8- —?cole Polytechnique F?d?rale de Lausanne10.13039/501100001703
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCatalytic Alkyne Reactions · Synthetic Organic Chemistry Methods · Asymmetric Synthesis and Catalysis
Propargylic alcohols 1, readily accessible from terminal alkynes and carbonyl compounds, are versatile and widely used building blocks in organic synthesis. ?−? ? The combination of a CC bond with a vicinal hydroxyl group makes these substrates particularly susceptible to activation by Brønsted acids, Lewis acids, and transition metals, thereby enabling the numerous powerful synthetic transformations. They also serve as precursors to other important synthons. For instance, two competing reactions, namely, the Meyer–Schuster rearrangement (Schemea)? and the Rupe rearrangement (Schemeb),? convert 1 into two isomeric α,β-unsaturated carbonyl compounds 2 and 3, respectively, depending on the reaction conditions and the nature of substituents on the propargylic alcohols. Under classical acidic conditions, the Meyer–Schuster reaction has mainly been limited to substrates lacking β-hydrogens, rendering the Rupe pathway inaccessible. ?,?
Mechanistically, allenic cation 4 has been proposed as the key intermediate in the Meyer–Schuster reaction. Trapping this highly reactive cationic species with a variety of nucleophiles has enabled the development of numerous synthetically powerful transformations.? On the other hand, reaction conditions that halt the Rupe rearrangement at intermediate 1,3-enyne 5, which is another class of important building blocks in organic synthesis, have been identified. ?−? ? ? ? Exploiting the reactivity of the enyne toward radical addition, Bao and Li developed an elegant iron-catalyzed domino dehydrative alkylation of propargylic alcohols to access functionalized 1,3-enynes (Schemec).?
The conversion of propargylic ethers to enynes under basic conditions has been only sporadically reported. Notably, Arens and co-workers showed that the treatment of propargylic ethers 6 with sodium amide in liquid ammonia afforded enynes 7 in good yields. ?,? However, the scope of this reaction has not been systematically investigated.? In connection with our ongoing project,? we had occasion to examine the carbocyclization of 8 via its zinc enolate? and observed the formation of 1,3-enyne 9 as a minor product, rather than the anticipated cyclization product. This unexpected outcome, combined with the facile accessibility of propargylic ethers and the synthetic value of 1,3-enynes, ?−? ? ? motivated us to investigate this transformation in detail. Herein, we report that exposure of a diethyl ether solution of propargylic ethers 10 to LDA (3.0 equiv) affords terminal 1,3-enynes 11 in high yields with good E selectivity. When 4 equiv of LDA was employed, the in situ-formed lithium acetylide can be trapped by electrophiles to generate functionalized 1,3-enynes 12 (Schemec). Importantly, the relative positions of the alkene and alkyne units in 1,3-enynes 11 and 12 differ from those obtained in the transformation reported by Arens and co-workers (cf. compound 7).?
Trimethyl[(2-phenylpent-3-yn-2yl)oxy]silane (10a) (R^1^ = Me; R^2^ = Ph) was selected for reaction optimization (Supporting Information). Some key experimental observations emerged. (a) Three equivalents of LDA proved to be optimal. LTMP (lithium 2,2,6,6-tetramethylpiperidide) gave a slightly lower yield of 11a under otherwise identical conditions, whereas no product was formed when nBuLi and NaH were employed as bases. (b) The presence of ZnBr_2_ and InBr_3_ was detrimental to the desired reaction. (c) Among the hydroxyl protective groups examined (OPiv, OBz, and OBoc), the OTMS group provided superior results. (d) Comparable outcomes were obtained in diethyl ether and tetrahydrofuran (THF), with an optimal substrate concentration of 0.15 M in Et_2_O. Overall, addition of LDA (3.0 equiv) to a diethyl ether solution of 10a (c = 0.15 M) at −78 °C, followed by warming to room temperature and stirring for 45 min, afforded (E)-4-phenyl-pent-3-en-1-yne (11a) in 74% yield. The E geometry of the double bond was assigned to the major product on the basis of the NOE experiment.
With the optimized conditions in hand, the reaction scope was examined starting with the TMS ether of the tertiary propargylic alcohols (Scheme). The TMS ether of 2-(4-hydroxyphenyl)pent-3-yn-2-ol (10b) was converted into 11b in 95% yield. The phenolic hydroxyl group was tolerated, although 4.5 equiv of LDA was required to ensure complete conversion. The TMS ether of 2-(pyridin-4-yl)pent-3-yn-2-ol (10c) was similarly transformed into 11c, demonstrating compatibility with a basic heteroarene. The presence of an aryl substituent in 10 was not a prerequisite, since 1,1-dialkyl-substituted but-2-yn-1-ol derivatives were converted into the corresponding 1,3-enynes (11d–11i) in excellent yields. A diyne substrate (10j) was highly regioselectively converted into enediyne 11j. Cyclic ketone-derived propargylic alcohols, including those originating from cyclohexanones (11k–11n), tetrahydro-4H-pyran-4-one (11o), N-Boc-piperidin-4-one (11p), N-Boc-2-azaspiro[3,5]nonan-7-one (11q), and adamantan-2-ones (11r and 11s), were smoothly converted into the corresponding enynes. Chiral substrates delivered enynes 11t and 11u without detectable epimerization. In addition, a range of prop-2-yn-1-ylidenecycloalkanes (11v–11z) were obtained in good yields. Importantly, diverse functional groups, including silyl ether, alkyl azide, chloride, methyl ether, acetal, and carbamate, were well tolerated under these reaction conditions.
We next investigated the conversion of the TMS ethers derived from secondary propargylic alcohols (Schemeb). Treatment of TMS ether of tridec-2-yn-4-ol (10aa) with LDA (3.0 equiv) in Et_2_O afforded tridec-3-en-1-yne (11aa) in 52% yield (1.7:1 E:Z). Notably, the regioisomeric tridec-1-en-3-yne (13 (inset of Schemeb)) was not detected. Enynes 11ab–11ae were similarly prepared as a mixture of E and Z isomers.
A gram-scale experiment converted 10d (1.0 g, 3.23 mmol) into 11d in 90% yield, highlighting the practicality and scalability of this protocol.
We note that no reaction occurred when the TMS ether of 2-phenylhept-3-yn-2-ol was subjected to the standard conditions. Therefore, the protocol can be applied only to the synthesis of terminal alkynes. To circumvent this limitation, functionalization of the terminal alkyne generated in situ was subsequently explored. As shown in Scheme, increasing the LDA loading to 4 equiv enabled trapping of the resulting alkynyllithium with a variety of electrophiles, including carbon dioxide, dimethyl carbonate, gaseous formaldehyde, trimethylsilyl chloride, and S-methylmethanethiosulfonate,? to afford functionalized internal enynes 12a–12g in excellent yields.?
Control experiments were conducted to gain insight into the reaction mechanism. When propargylic alcohol 10d was subjected to the standard conditions and the reaction was quenched with D_2_O, C1-deuterated product 11d-D and nondeuterated 11d were obtained in 80% and 15% yields, respectively (Schemea). No C3-deuterated products were detected in the reaction mixture. In contrast, exposure of 10d-D 3 to the standard conditions afforded a mixture of four identifiable compounds, consisting of nondeuterated 11d together with deuterated products 14–16 (Schemeb). Notably, the C3 vinylic atom was deuterated in products 14–16. For the success of this deuterium labeling experiment, it is important to generate LDA with an excess of nBuLi to ensure that the diisopropylamine (iPr_2_NH) formed in situ was fully reconverted into LDA, thereby minimizing competitive protonation of organolithium intermediates. Reaction of 11g, obtained from 10g under the standard conditions, with LDA furnished cyclopropane derivative 17 in 80% yield. By contrast, subjecting 10g to a large excess of LDA (9.0 equiv) led to a mixture of two inseparable compounds, biscyclopropane 18 and 1,2-disubstituted cyclopentene 19, in a 54% combined yield (1.4:1). Finally, reaction of 10d with 1.0 equiv of LDA, followed by quenching at approximately 30% conversion, allowed the isolation of cumulene 20 in ca. 1% yield.? Subsequent reaction of 20 with 1.3 equiv of LDA delivered expected 1,3-enyne 11d in 80% yield. Collectively, these control experiments support the involvement of a cumulene intermediate as well as both C1-lithiated and C3-lithiated species in the conversion of 10 into 11.
Building on the control experiments presented above, a plausible reaction mechanism is proposed in Scheme. Deprotonation of 10, followed by elimination of lithium trimethylsilyl oxide, would generate cumulene 20, which would then undergo further deprotonation to furnish 21. Given that the acidity of allene C(sp^2^)–H bond is comparable to that of an acetylene C–H bond (pK a ∼ 25),? resulting organolithium species 21 could not be protonated by iPr_2_NH (pK a ∼ 35). Instead, it underwent isomerization to give vinyl lithium species 22 (pK aH ∼ 42), which could subsequently be protonated by iPr_2_NH to afford 1,3-enyne 11. Once formed, 11 would be immediately deprotonated by LDA leading to lithium acetylide 23. Quenching the reaction mixture with water would then deliver product 11, whereas trapping with an electrophile would lead to the formation of internal alkyne 12. Note that the acidity of the C(sp)–H bond in 11 is significantly higher than that of the propargylic C(sp^3^)–H bond in starting material 10. As a result, 11 is deprotonated before the complete consumption of 10. Consequently, 3 equiv of base is required to drive the reaction to completion. This mechanistic scenario is consistent with all experimental observations shown in Scheme.
In summary, we have developed a base-mediated conversion of the TMS ether of propargylic alcohols into 1,3-enynes. The transformation proceeds under mild conditions and tolerates a wide range of functional groups. Given the ready accessibility of the starting materials, this method provides a practical and attractive alternative to 1,3-enynes of significant synthetic importance.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Xue Z.-H.Liu Q.-H.Liu G.Sun Z.Qin T.Bian S.-W.Han Y.-P.Liang Y.-M.Recent Synthetic Transformation of Propargylic Alcohols Asian J. Org. Chem.202514 e 7020910.1002/ajoc.70209 · doi ↗
- 2Chao J.Yang R.Huang J.Chen X.Song X.-R.Xiao Q.Advances in the Direct Transformations of Propargylic Alcohols and Sulphur-Containing Reagents Org. Biomol. Chem.2025238862889010.1039/D 5OB 01220 A 40965182 · doi ↗ · pubmed ↗
- 3Beluze C.Hu T.Bouyssi D.Monteiro N.Amgoune A.Metallaphotoredox Reactions of Propargyl Alcohol Derivatives: New Perspectives on Allene Synthesis Synthesis 2025572935294510.1055/a-2655-3710 · doi ↗
- 4Meyer K. H.Schuster K.Umlagerung Tertiärer Äthinyl-Carbinole in Ungesättigte Ketone Ber. Dtsch. Chem. Ges. A/ B 19225581982310.1002/cber.19220550403 · doi ↗
- 5Rupe H.Kambli E.Ungesättigte Aldehyde Aus Acetylen-Alkoholen Helv. Chim. Acta 1926967267210.1002/hlca.19260090185 · doi ↗
- 6Engel D. A.Dudley G. B.The Meyer–Schuster Rearrangement for the Synthesis of α,β-Unsaturated Carbonyl Compounds Org. Biomol. Chem.200974149415810.1039/b 912099 h 19795050 · doi ↗ · pubmed ↗
- 7Swaminathan S.Narayanan K. V.Rupe and Meyer-Schuster Rearrangements Chem. Rev.19717142943810.1021/cr 60273 a 001 · doi ↗
- 8Zhu Y.Sun L.Lu P.Wang Y.Recent Advances on the Lewis Acid-Catalyzed Cascade Rearrangements of Propargylic Alcohols and Their Derivatives ACS Catal.201441911192510.1021/cs 400922 y · doi ↗