Regularized Single‐Cell Imaging Enables Generalizable AI Models for Stain‐Free Cell Viability Screening
Pan Deng, Deasung Jang, Samuel G. Berryman, Simon P. Duffy, Hongshen Ma

TL;DR
A new imaging method helps train AI models to accurately assess cell viability without stains, working across different cell types and treatments.
Contribution
Regularized single-cell imaging in nanowells improves AI model generalizability for stain-free cell viability screening.
Findings
The model accurately identified live and dead cells for unseen compounds and cell types.
Non-destructive brightfield imaging allows kinetic studies of cell viability over time.
Dose-response curves matched fluorescence assays despite limited training data.
Abstract
Cell viability assays are essential tools in biomedical research and drug development. Artificial intelligence (AI) offers the potential to simplify these assays by predicting cell viability directly from brightfield microscopy images, but current models lack generalizability across diverse cell types and treatments. Here, we introduce a strategy called “regularized imaging”, where single cells are isolated in nanowells to generate standardized image patches that simplify segmentation and improve training data quality. We trained our model using example images of live and dead cells from a single cell line exposed to four cytotoxic conditions (ethanol, andrographolide, daunorubicin, and serum starvation). Despite this narrow training dataset, the resulting model accurately identified live and dead cells after treatments by previously unseen compounds, successfully replicating…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
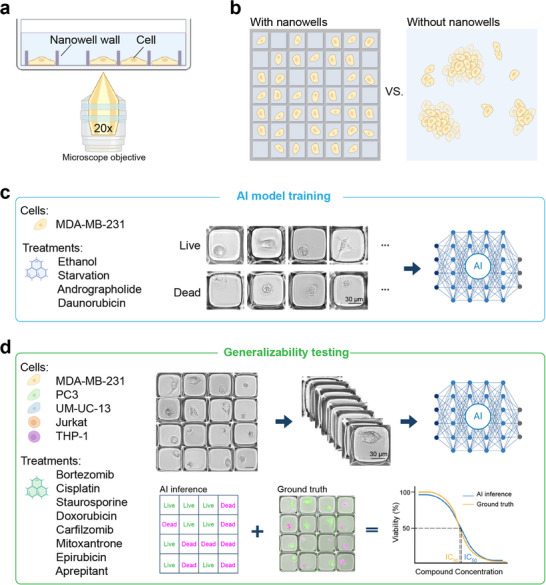 FIGURE 1
FIGURE 1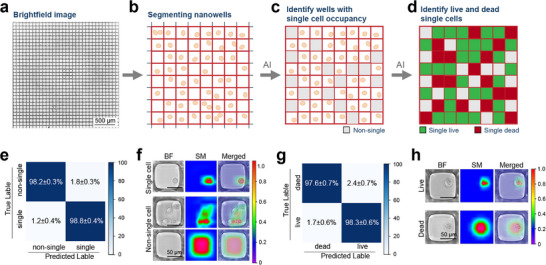 FIGURE 2
FIGURE 2 FIGURE 3
FIGURE 3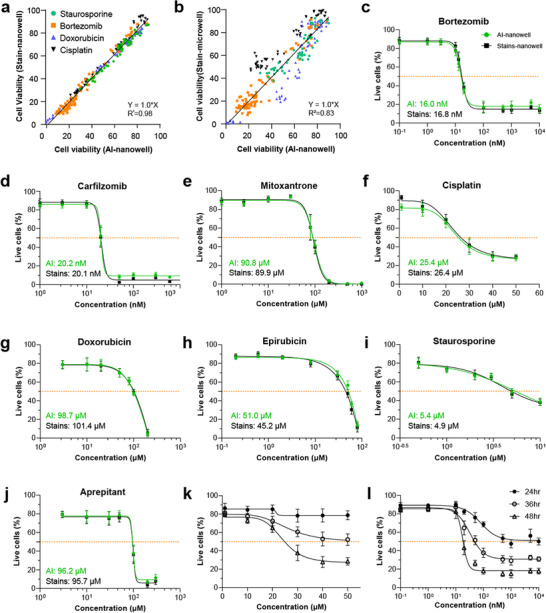 FIGURE 4
FIGURE 4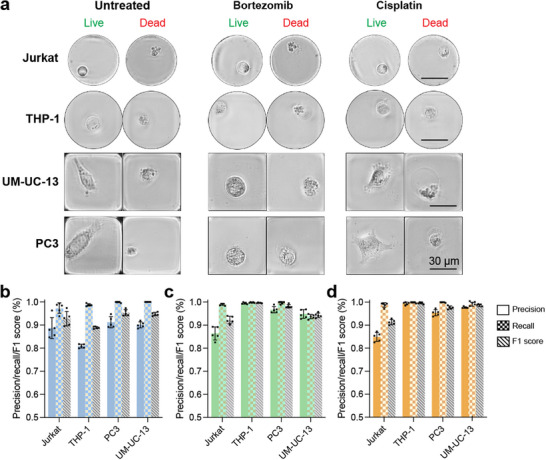 FIGURE 5
FIGURE 5| 1st AI model | 2nd AI model | |||
|---|---|---|---|---|
| Treatment | Single cell | Non‐single cell | Live single cell | Dead single cell |
| Culture media | 18 078 | 17 401 | 17 772 | 309 |
| Culture media‐no FBS | 14 426 | 15 638 | 8654 | 8922 |
| Ethanol | 18 005 | 19 970 | 170 | 17 921 |
| Andrographolide | 5383 | 3483 | 826 | 4567 |
| Daunorubicin | 30 351 | 29 751 | 15 831 | 11 534 |
- —Natural Sciences and Engineering Research Council of Canada10.13039/501100000038
- —China Scholarship Council10.13039/501100004543
- —Tai Hung Fai Charitable Foundation
- —Society for Laboratory Automation and Screening Graduate Education Fellowship
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCell Image Analysis Techniques · Single-cell and spatial transcriptomics · Andrographolide Research and Applications
Introduction
1
Cell viability screens are essential assays used to evaluate the activity of chemical compounds on target cells in biomedical research and drug development. Traditional assays, such as MTT, SRB, and CellTiter‐Glo, measure metabolic activity to obtain a bulk‐level measure of cell viability using light absorption, fluorescence or luminescence, which can be readout using standard microplate readers [1, 2]. While widely used, these methods lack single‐cell resolution and require sampling consistent numbers of homogeneous cells to produce reliable results. Fluorescence‐based live/dead staining enables viability screening at the single‐cell level using microscopy or flow cytometry [3]. However, these approaches require additional staining and incubation steps, increasing complexity, cost, and assay time. Moreover, imaging stained cells using fluorescent microscopy could reduce cell viability due to phototoxicity [4, 5, 6], which limits these assays to single‐time‐point measurements and prevents the acquisition of kinetic data to assess drug response dynamics.
Recent advances in artificial intelligence (AI) have shown the possibility for stain‐free viability prediction directly from microscopy images [7, 8, 9, 10, 11, 12, 13]. These approaches typically fall into two categories: object classification, where models are trained on image patches centered around individual cells [8, 9, 10, 14], or pixel classification, where convolutional neural networks based on the U‐Net architecture are used to generate pixel‐level segmentation masks across entire microscopy fields [11, 12, 15, 16, 17]. While promising, current models suffer from poor generalizability—they must be re‐trained on each new cell type and compound in order to achieve their predictive accuracy. A truly generalizable model that performs reliably on previously unseen cell types and compounds has not been demonstrated. This lack of generalizability severely limits the utility of AI‐based viability assays, where retraining for each new condition would negate the benefit of a stain‐free approach.
One of the key challenges of previous approaches for AI‐based cell viability assays is single‐cell segmentation, which is required to isolate features indicative of cell viability during training and to generate a single‐cell readout during inference. Manual cropping of single cells into individual image patches is labor‐intensive and impractical for large datasets [18, 19]. While classical computer vision methods attempt to automate this cropping process, they require cells to be seeded at very low densities to minimize contamination from neighboring cells [14]. Several AI‐based tools have been developed to segment cells based on cell boundaries [20, 21]. However, these methods often perform poorly in detecting dead cells, particularly when dead cells are in contact with live cells or with each other. Therefore, there is a critical need for an approach that enables robust single‐cell isolation and segmentation across diverse cell types while enabling microscopy observation of cellular features necessary for viability assessment.
Here, we report on an approach to overcome the generalizability challenge in AI‐based cell viability assays by isolating single cells in nanoliter wells (nanowells) integrated into standard glass‐bottom microwell plates. This strategy, which we term “regularized imaging”, simplifies cell segmentation and normalizes single‐cell images to enable AI models to learn morphological features specific to live and dead cells. We demonstrate that this approach accurately reproduces IC_50_ curves of fluorescence‐based assays for compounds not used in the training data set, as well as provides generalizable live/dead detection on previously unseen cell types. Our results show that cell viability can be associated with conserved morphological signatures that can be learned and generalized by AI using regularized imaging.
Results
2
Approach
2.1
Our approach involves isolating single cells into nanowells integrated into standard glass‐bottom imaging microwells to obtain regularized microscopy images of live and dead cells under diverse experimental conditions (Figure 1a). Isolating single cells in nanowells significantly simplifies cell segmentation, enabling the acquisition of unambiguous training data based on distinct cellular characteristics (Figure 1b). The training data set is collected on live and dead MDA‐MB‐231 cells. Cell death was induced using four conditions, which include exposure to ethanol, andrographolide, daunorubicin, and serum starvation. Brightfield images of single cells were labeled based on cell viability determined using fluorescence live/dead staining, which subsequently served as ground truth data for training the AI model (Figure 1c). Following training, we assessed the generalizability of the model in determining cell death resulting from exposure to previously unseen anticancer agents at various concentrations. Using viability predictions from the AI model, we generated dose‐response curves and calculated the IC_50_ value for comparison with conventional fluorescence‐based assays. Additionally, we demonstrate the utility of this approach to perform repeated cell viability analysis to characterize the response dynamics of anticancer agents. Finally, we evaluated the generalizability of our model to predict the viability of previously unseen cells, including both adhesion and suspension cells, highlighting the broad utility of this approach for drug screening workflows (Figure 1d).
Study overview. (a) Microscopy using nanowell‐in‐microwell plates. (b) Imaging with and without nanowells. (c) AI model training using MDA‐MB‐231 cells treated with ethanol, serum starvation, andrographolide, and daunorubicin were used to generate examples of live and dead cell images. (d) Evaluating the generalizability of the trained AI model on previously unseen cell types and compounds. Cells in nanowells were imaged, segmented, and analyzed by the AI model. The AI inference results are compared against ground truth obtained via live/dead cell staining to reconstruct IC50 curves.
The nanowell microstructures are fabricated on glass substrates and subsequently integrated with plastic microwell frames to create nanowell‐in‐microwell plates. This fabrication process has been described earlier [22, 23]. Briefly, the nanowells are formed using photolithographic patterning of a polyurethane‐based UV curable polymer on rectangular glass substrates. This process involves spin‐coating the prepolymer on a glass slide, followed by UV exposure through a photomask to create nanowells with dimensions 70 × 70 × 60 µm (length × width × height), resulting in an open‐top chamber with a volume of 0.29 nL. A plastic frame conforming to ANSI‐standard 384‐well plate dimensions is then adhered to each glass slide resulting in nanowells‐in‐microwell plate, where each microwell contains ∼1200 nanowells.
Single Cell Image Acquisition and Preparation
2.2
To acquire training and testing data, cells treated with cytotoxic agents were randomly seeded into nanowells at a low density, at 35%–45% of the number of nanowells. The nanowell‐in‐microwell plates were imaged in brightfield and fluorescence using an inverted microscope with a 20 × phase contrast objective (Figure 2a). Each microscopy field is segmented into single nanowell image patches of 248 × 248 pixels using a custom OpenCV script (Figure 2b). Briefly, the raw images are preprocessed with thresholding and morphological transformations for contour detection. The contours with areas close to the nanowell size are identified as nanowell boundaries. Finally, nanowells are extracted by cropping around the centroid of each identified contour, resulting in 248 × 248‐pixel image patches. The single nanowell image patches are first analyzed to identify nanowells that contain only single cells. This analysis is performed by training an AI model to classify the image patches as those containing single cells or non‐single cells (0 or ≥2 cells per nanowell) (Figure 2c). To generate the training data for this model, we manually labeled 137,988 single nanowell images based on those two categories. The labeled dataset was used to train an Xception convolutional neural network (CNN) model, which was pre‐trained on the ImageNet database to leverage its ability to detect features such as edges, shapes, textures, and patterns relevant to cell imaging. The model's input layer was adjusted to accommodate the dimension of the nanowell image patches. Additional custom layers were integrated to adapt the model for binary classification of nanowell images. Evaluating the performance of the model using five‐fold cross‐validation on the labeled dataset achieved a training accuracy of 99.3 ± 0.4% and a validation accuracy of 99.0 ± 0.3%. Testing across validation splits on 27,597 unseen images achieved an average classification accuracy of 98.8 ± 0.4% for identifying nanowells containing only a single cell (Figure 2e). Saliency maps for two classes were focused on cells, or the lack of cells, suggesting the classification was made on relevant data (Figure 2f).
Data processing and analysis. (a–d) Image acquisition and analysis pipeline. (a) Acquisition of brightfield (BF) microscopy images. (b) Segmented into single nanowell image patches. (c) Use an AI model to identify nanowells occupied by a single cell. (d) Use another AI model to assess single‐cell viability. (e) Confusion matrix for the single cell occupancy AI model. (f) Representative saliency maps for the single cell occupancy AI model. (g) Confusion matrix for the cell viability AI model. (h) Representative saliency maps for the cell viability AI model.
AI Model for Cell Viability Analysis
2.3
To train an AI model for cell viability analysis from microscopy images, we acquired a training set of 86,506 image patches of single MDA‐MB‐231 cells in nanowells. Ground truth was obtained by staining these cells using fluorescence live/dead stains. Cell death was induced in these cells by exposure to ethanol, andrographolide, daunorubicin, or serum starvation. These treatments are designed to induce a wide range of cell deaths, including apoptosis (ethanol, andrographolide, daunorubicin) [24, 25], necrosis (ethanol) [24], autophagy (ethanol, serum starvation, andrographolide) [26, 27, 28], mitochondrial stress (ethanol) [29], cell cycle arrest (andrographolide) [30], DNA damage (daunorubicin) [31], and nutrient deficiency (serum starvation) (Figure S1, Table S1).
An adapted Xception CNN model was employed to classify 248 × 248‐pixel images using fluorescence microscopy results as ground truth labels (Figure 2d). We evaluated the performance of this model using five‐fold cross‐validation on 69,206 images, which achieved an accuracy of 99.5 ± 0.3% for training and 98.9 ± 0.5% for validation. We then tested the model developed from each cross‐validation split on 17,300 unseen images. The model achieved a classification accuracy of 97.6 ± 0.7% for dead cells and 98.3 ± 0.6% for live cells (Figure 2g). Saliency map confirmed that model predictions were predominantly based on cellular morphology rather than background regions (Figure 2h).
Generalizability to Previously Unseen Chemical Compounds
2.4
To investigate the generalizability of our AI model to determine cell death resulting from exposure to previously unseen chemical compounds, we acquired imaging data on MDA‐MB‐231 cells treated with eight additional anticancer agents with different death mechanisms: bortezomib, carfilzomib, cisplatin, staurosporine, doxorubicin, mitoxantrone, epirubicin, and aprepitant (Table S1). As before, cells were incubated in nanowell‐in‐microwells and exposed to a broad range of concentrations of each compound. After 24 or 48 h of incubation, cells were stained using a fluorescence live/dead stain and subsequently imaged in brightfield and fluorescence (Figure 3). Cell viability was then assessed using our trained AI model, as well as conventional fluorescence imaging. Our results show that the AI‐based viability measurements closely matched results obtained using standard fluorescence staining, achieving an R^2^ value of 0.98 (Figure 4a).
Example brightfield images of live and dead MDA‐MB‐231 cells in nanowells after treatment using various compounds.
Generalizability to previously unseen compounds. (a) Correlation for viability determined using AI‐inferencing versus fluorescence live/dead staining on the same aliquots of MDA‐MB‐231 cells in nanowells (R2 = 0.98). (b) Correlation for viability determined using AI‐inferencing of MDA‐MB‐231 cells in nanowells versus fluorescence live/dead staining on different aliquots assess in standard microwells (R2 = 0.83). (c–j) Dosage response curves for MDA‐MB‐231 cells in nanowells after exposure to eight previously unseen compounds obtained using AI‐inferencing and fluorescence live/dead cell staining. IC5₀ concentrations are assessed at the half‐maximal cell death level (indicated by orange dashed lines) (n = 9). (k–l) Dosage response curves for MDA‐MB‐231 cells in nanowells after exposure to cisplatin (k) and bortezomib (l) (n = 9). All dosage response data are presented as mean ± standard deviation of three independent experiments performed in triplicate.
To determine if AI‐based viability screening in nanowells could reliably substitute fluorescence‐based assays performed in standard microwells, we tested separate aliquots of the same cells for each treatment condition in conventional imaging microwells without nanowells. Comparing cell viability determined using AI‐based viability screening in nanowells to fluorescence‐based screening in microwells revealed a strong correlation (R^2^ = 0.83) with slightly greater, but unbiased, variability expected from the testing of distinct aliquots (Figure 4b). Similar differences between cells cultured in microwells and nanowells have been reported previously [32], including in 96‐well plates, 384‐well plates, and microfluidic chambers. These differences are likely attributable to reduced cell motility, limited cell–cell communication, and altered local concentrations of drugs and signaling molecules.
To further assess whether our AI model has learned the morphology associated with cell death without being limited to nanowell substrates, we used our AI model to assess cell death on images of single cells cultured in standard microwells (384‐well plates). These cells are cultured at low density and have been cropped to the same size as the nanowell images. We evaluated cells exposed to Bortezomib, Doxorubicin and Cisplatin and found the model could assess live cells with 96%–99% accuracy, and dead cells with 92%–93% accuracy (Figure S3). These results confirm that the model can perform generalized analysis of cell death independent of the imaging substrate. Together, these results demonstrate that AI‐powered viability assessment provided results comparable to fluorescence staining, suggesting they can effectively replace fluorescence‐based cell viability assays.
To investigate the utility of our AI model for generating accurate dose‐response curves suitable for screening applications, we acquired dose‐response data for all 8 compounds using both AI‐based and fluorescence‐based screening (Figure 4c–j). IC_50_ concentrations obtained using both two methods were highly concordant, with an average deviation of only 4.5% (range: 0.5%–12.8%). Collectively, these results confirm that our AI‐based screening method can reliably determine IC_50_ values for novel compounds, demonstrating its potential integration into routine screening workflows.
Kinetic Drug Response
2.5
An important advantage of our AI‐based cell viability assay is its non‐destructive nature, which enables repeated measurements at multiple time points. To demonstrate this capability, we performed time‐resolved viability profiling using our AI‐based method to measure the dynamic dose‐response of MDA‐MB‐231 cells treated with cisplatin and bortezomib (Figure 4k,l). Our analysis revealed a clear, time‐dependent reduction in cell viability at 24, 36, and 48 h of incubation, with bortezomib showing more rapid cytotoxic activity compared to cisplatin. These results demonstrate the potential to use AI‐based cell viability analysis in kinetic studies to distinguish between fast‐acting and slow‐acting compounds, which is very difficult to perform using traditional viability assays.
Generalizability to Previously Unseen Cells
2.6
To investigate whether our AI model has learned morphologies specifically associated with cell death that could be generalized to other cells, we used our model to analyze cells from four previously unseen cell lines: Jurkat, THP‐1, UM‐UC‐13, PC‐3 (Figure 5). Jurkat and THP‐1 are suspension cell lines derived from lymphoblastic and monocytic leukemia, while PC3 and UM‐UC‐13 are adherent cell lines derived from prostate and bladder cancer. Suspension cells display round morphologies, whereas adherent cells display spindle‐like and sheet‐like morphologies similar to MDA‐MB‐231 cells. Cell sizes also varied considerably: microscopy measurements showed that MDA‐MB‐231 (area = 678 ± 224 µm^2^), PC3 (722 ± 171 µm^2^), and UM‐UC‐13 (422 ± 84 µm^2^) were significantly larger compared to Jurkat (106 ± 33 µm^2^) and THP‐1 (184 ± 35 µm^2^). Images of live cells were acquired from culture, while cell death was induced using bortezomib and cisplatin. Despite significant differences in cell morphology, our AI‐model was able to identify live and dead cells with high accuracy when compared with ground‐truth labels from live/dead staining. Precision, recall, and F1 scores averaged across all four cell lines were 0.92 ± 0.06, 0.99 ± 0.06, and 0.95 ± 0.04, respectively (Figure 5b–d). Expectedly, Jurkat cells had the lowest performance with precision, recall, and F1 scores of 0.87 ± 0.03, 0.97 ± 0.02, and 0.93 ± 0.02, respectively. This result is likely attributable to the model being trained only on with MDA‐MB‐231 adherent cell lines and the smaller size of Jurkat cells, which reduced the available pixels for the AI model to analyze.
Generalizability to previously unseen cell types. (a) Example brightfield images of live and dead Jurkat, THP‐1, UM‐UC‐13, and PC3 cells after treatment with bortezomib and cisplatin. (b–d) Precision, recall, and F1 score of the AI‐inferencing for live cells in culture medium (b), dead cells induced using bortezomib (c), and dead cells induced using cisplatin (d). Data points are presented as mean ± standard deviation of five independent wells (n = 5).
Discussion
3
This study presented a generalizable AI‐powered assay capable of determining cell viability directly from microscopy images without needing to chemically or fluidically manipulate the sample. Our approach leveraged regularized imaging of single cells in nanowells, which significantly simplified cell segmentation and enabled the training of generalizable AI models to assess cell viability. Critically, despite focused training using example images of live and dead cells from a single cell line subjected to four cytotoxic conditions (ethanol, andrographolide, daunorubicin, and serum starvation), our model accurately assessed cell viability over a much larger set of previously unseen chemical treatments, effectively reproducing dose‐response curves obtained using fluorescence‐based assays. Moreover, our model demonstrated remarkable robustness across diverse cell types, including both adherent and suspension cells. Additionally, the non‐destructive nature of this assay enables repeated measurements to provide kinetic response data for detailed pharmacological characterization.
Previous AI‐based viability studies often image cells in traditional microwells, where migration and clustering complicate single‐cell segmentation. Low cell seeding densities can be used to mitigate this issue [14, 19], but this approach comes at the cost of throughput. Alternative single‐cell isolation approaches, such as droplet encapsulation [33, 34], often suffer from optical distortion caused by curved droplet interfaces, require complex cell handling steps, and preclude cell adhesion. Microarray‐based platform devices have simplified cell seeding through open‐well pipetting [35, 36, 37]. However, the work related to analyzing cell viability used fluorescent labeling rather than intrinsic cellular morphology to distinguish live and dead cells. This is likely driven by non‐ideal imaging substrates, such as thick PDMS layers or uneven etched glass, that degrade image quality. In contrast, our nanowell features an optically flat and thin glass bottom that supports both adherent and suspension cells while enabling high‐resolution microscopy. By physically isolating individual cells in nanowells, it is possible to obtain optically clean images that preserve single‐cell morphology without being confounded by neighboring cells. The availability of high‐quality training data enables the training of generalizable AI models that focus on intrinsic visibility‐related morphological features.
Compared with traditional viability assays (e.g., MTT, SRB and CellTiter‐Glow), our AI‐powered assay not only enables single‐cell‐level viability assessment, but also substantially streamlines drug screening workflows by eliminating the complexity and costs associated with assay incubation, fluorescence staining and imaging. Brightfield‐only imaging significantly improves throughput by reducing microscope camera exposure times and eliminating delays caused by switching between imaging channels during multi‐color fluorescence imaging [38]. Eliminating fluorescence imaging further reduces the cost of imaging equipment and staining reagents, which can be a barrier in large‐scale screening campaigns [39]. Importantly, the ability to repeatedly analyze the same sample to generate kinetic response profiles significantly reduces the number of cell aliquots needed for studies that must accommodate both fast‐ and slow‐acting compounds.
More broadly, our findings highlight the power of using regularized imaging in nanowells to train AI models to recognize biologically relevant cell morphologies that may be inaccessible to human perception. Classical cell morphology analysis methods, such as those employed by software like CellProfiler, rely on human interpretable features, which may lack sufficient complexity to detect nuanced morphological features indicative of specific biological states [40, 41]. Similarly, AI approaches that attempt to reconstruct live/dead stains typically focus on the regions highlighted by the fluorescent signal, which may overlook generalizable features that are informative for assessing cell viability across multiple compounds and cells [12]. In contrast, regularized imaging using nanowells allows cell images to be directly associated with the assay outcomes to provide standardized, high‐quality training datasets that encourage AI models to learn features relevant to the intended biological outcomes. These features could be pixel‐level patterns that cannot be captured as human‐interpretable descriptions, effectively overcoming limitations imposed by human language and cognition. We anticipate that this approach can be used to train AI models to identify cell morphologies associated with challenging cell phenotypes, states, and properties, thereby significantly streamlining assays to detect these cells at the single cell level.
Methods
4
Nanowell Fabrication
4.1
A glass slide (75 × 50 × 0.3 mm, Abrisa Technologies) substrate was washed with acetone (Sigma‐Aldrich) and isopropyl alcohol (IPA, Sigma‐Aldrich) for 20 min each. After drying, the slide underwent plasma cleaning for 2 min and was then treated with 10% v/v TMSPMA (M6514, Sigma‐Aldrich) in ethanol at 70°C for 2 h. The slide was rinsed with and baked at 80°C for 1 h. A layer of UV‐curable prepolymer was spin‐coated onto the treated slide and nanowell patterning was achieved by a photolithographic process which involves a UV light (20 mW cm^−2^) exposure through a photomask with nanowell features. The slide was rinsed with IPA to remove uncured prepolymer, resulting in a bas‐relief structure of nanowells on the glass surface. Finally, the structured glass was bonded to the bottom of a standard 384‐well plate frame (Product No. 206384, Grace Bio Labs) to create a nanowell‐in‐microwell plate. Each microwell contained approximately 1,200 nanowells, with individual nanowells measuring 70 × 70 × 60 µm (length × width × height). Jurkat cells, which are significantly smaller than adherent cells, were imaged in prototype circular nanowells (diameter = 55 µm) provided by ImageCyte Technologies (Vancouver, Canada).
Chemical Components
4.2
Andrographolide (Product No. 365645, Sigma‐Aldrich), daunorubicin hydrochloride (Product No. 30450, Sigma‐Aldrich), Ethanol (Sigma‐Aldrich), bortezomib (Product No. 5043140001, Sigma‐Aldrich), carfilzomib (Product No. S2853, Selleckchem), cisplatin (Product No. S1166, Selleckchem), staurosporine (Product No. S4400, Sigma‐Aldrich), doxorubicin hydroxydaunorubicin (Product No. S1208, Selleckchem), mitoxantrone dihydrochloride (Product No. M6545, Sigma‐Aldrich), epirubicin hydrochloride (Product No. S1223, Selleckchem), aprepitant (Product No. S1189, Selleckchem).
Cell Culture
4.3
MDA‐MB‐231 (HTB‐26, ATCC), UM‐UC‐13 (a kind gift from Dr. Peter Black, University of British Columbia, Vancouver, Canada), and PC3 (CRL‐1435, ATCC) were cultured in DMEM medium (Product No. 11965092, Life Technologies) supplement with 10% fetal bovine serum (FBS, Product No. F4135, Sigma‐Aldrich) and 1% penicillin‐streptomycin (Product No. 15140122, Life Technologies). Jurkat (TIB‐152, ATCC) and THP‐1 (TIB‐202, ATCC) were cultured in RPMI 1640 medium (Product No. 11835030, Sigma‐Aldrich) supplement with 10% FBS and 1% penicillin‐streptomycin. All cells were cultured in a humidified incubator with 5% CO_2_ at 37°C.
Cell Treatment Inside Nanowells
4.4
Adhesion cells (MDA‐MB‐231, UM‐UC‐13, and PC3) were washed in phosphate‐buffered saline (PBS, Product No. 10010001, Life Technologies), detached from culture flasks (Product No. 156367, Thermo Scientific) using 0.05% Trypsin/EDTA (Product No. 25300062, Life Technologies), and resuspended in culture media with 10% FBS, except for MDA‐MB‐231 cells, which were resuspended in media containing 5% FBS. Suspension cells were washed once in their culture media before drug treatment. To initiate a drug treatment, drugs diluted with culture media were added to the cell suspension and thoroughly mixed in a centrifuge tube to achieve the desired final drug concentration. The resulting cell‐drug mixture was pipetted into a nanowell‐in‐microwell plate at a density of approximately 500 cells per microwell. After centrifuging the cells down to the nanowell bottom, the whole plate was put into the cell culture incubator for a predetermined treatment period.
MDA‐MB‐231 cells were treated with the indicated concentrations of cisplatin, bortezomib, epirubicin, doxorubicin, aprepitant, and staurosporine for 48 h, and with mitoxantrone for 24 h Jurkat cells were treated with bortezomib (150 nM) and cisplatin (50 µM) respectively for 24 h THP‐1 cells were treated with bortezomib (1 µM) and cisplatin (50 µM) respectively for 24 h. UM‐UC‐13 were treated with bortezomib (200 µM) and cisplatin (80 µM) respectively for 24 h PC3 cells were treated with bortezomib (100 µM) for 36 h and cisplatin (150 µM) for 24 h Untreated conditions were conducted in cells cultured in their complete culture medium for 24 h.
Viability Staining and Image Acquisition
4.5
Cells were seeded into nanowells of the 384‐well nanowell‐in‐microwells plate at a ratio of 40% and brightfield images were acquired using an inverted phase‐contrast microscope (ECLIPSE Ti2 Series, Nikon). This microscopy system had an integrated perfect focusing system to enable scanning a whole microwell in ∼2s. Cells were imaged using a 20 × phase contrast objective (Plan Fluor 20 × Ph1 DLL, Nikon). Jurkat cells image using an additional 1.5 × magnification lens. All images were captured using a 16.25‐megapixel monochrome camera (DS‐Qi2, Nikon). Cells were subsequently stained inside nanowells using the LIVE/DEAD Cell Imaging Kit (Product No. R37601, Thermo Scientific) and imaged using mCherry (red) and EGFP (green) channels, following manufacturer's protocol. A cell was classified as live only if it satisfied all of the following criteria: (i) positive green fluorescence, (ii) cell size within ±30% of the mean for that cell type, and (iii) absence of visible membrane blebbing. Cells that did not meet all three criteria were classified as dead. These combined fluorescence‐ and morphology‐based labels served as the ground truth for live/dead staining in evaluating AI model predictions.
Automatic Nanowell Segmentation
4.6
Nanowells in brightfield images were segmented using OpenCV in Python, with each segmented nanowell image standardized to 248 × 248 pixels. Specifically, we first applied thresholding and morphological transformations to distinguish nanowells from other objects. These images were then processed using the findContours() function to extract contours. Contours with areas 50% smaller or larger than the expected nanowell area were filtered out. The centroids of the remaining contours were identified to mark the positions of detected nanowells. For square‐shaped nanowells, nanowell patches were extracted by cropping around the centroids to a size of 248 × 248 pixels. For round‐shaped nanowells, circular regions with a diameter of 248 pixels were cropped and subsequently zero‐padded to form square images (248 × 248 pixels).
AI Model Development
4.7
Two CNN models were developed to support this workflow. The first CNN model classified nanowells based on whether they contained a single cell (1 cell/nanowell) or non‐single cell (empty or >1 cell/nanowell). To ensure high‐quality segmentation, single cells that were out of focus were annotated as non‐single cells. For this model, we generated a training set of 137 988 nanowell images, where the ground truth for cell number was determined by manual checking. The second CNN model accepted only nanowells with single cells as input and classified the cells as either live or dead. The training set for this model included 69 176 nanowell images and used fluorescence LIVE/DEAD staining, cell size and absence of membrane blebbing as criteria for ground truth cell viability. To ensure that a diversity of cell death pathways was represented in the training dataset, we included MDA‐MB‐231 cells in either complete culture media, culture media without FBS, ethanol (5%–50% v/v), andrographolide (5–50 µM), or daunorubicin (0.1–100 µM). Cells were treated in nanowells for 4–48 h. The total available images were summarized in Table 1. Both CNN models were generated using a modified Xception deep learning model, implemented using Python 3.10 and TensorFlow 2.7. Specifically, the Xception model's input layer was modified to accept 3‐channel images with a dimension of 248×248 pixels. After the last convolutional layer (block14_sepconv2_act), the original classification head was replaced with a global average pooling layer, followed by a fully connected layer (1024 units, ReLU activation) and a final dense layer with 2 units and softmax activation. The modified model was compiled using the Adam optimizer with an initial learning rate of 0.001. Categorical cross‐entropy was used as the loss function to evaluate differences between predictions and actual labels, and model performance was evaluated with the accuracy metric. No learning rate scheduler was applied during training. The model was trained with a batch size of 32 for 15 epochs, using real‐time data augmentation through random vertical and horizontal flips during each epoch. We used five‐fold cross‐validation on 80% of the image dataset and the remaining 20% was used for testing. The final AI models were developed by training on the full dataset summarized in Table 1. Figure S2 shows the ROC curves, and the default threshold of 0.5 was used throughout our study. CNN model performance was evaluated by calculating precision, recall and F1 score for each microwell. The model training and testing were performed on the Digital Research Alliance of Canada's Graham computing cluster located at the University of Waterloo. The models were run on a GPU (NVIDIA V100 Volta) with 32 GB memory.
Model Validation on Unseen Cell Lines
4.8
The model performance was evaluated by the precision, recall and F1 score at a microwell level. For each treatment, the dominant class (live or dead) was defined as positive while the other one was defined as negative. The precision, recall and F1 score are determined as the following formula:
Generation of IC50 Curves
4.9
MDA‐MB‐231 cells were treated with eight previously unseen drugs. The concentration range was selected to span the known effective dose range. Following each prescribed incubation period, cells were first imaged using brightfield microscopy for AI‐based viability inference. Subsequently, cells were stained with the LIVE/DEAD Cell Imaging Kit and imaged via fluorescence microscopy to generate ground truth viability labels. Ground truth viability in each well was calculated as the proportion of live single cells relative to the total number of single cells. In parallel, we used the CNN model described above that had been trained against select conditions (complete culture, serum‐starvation, ethanol, andrographolide, and daunorubicin) to classify each single cell as live or dead, and to infer the percent viability of cells in the microwell. The IC_50_ value for each cell viability measurement was calculated based on the concentration of drug associated with 50% cell viability. The values were reported based on the mean of three independent experiments.
Author Contributions
H.M. supervised the study. H.M. and P.D. conceived the idea. P.D., D.J., and S.G.B. performed the experimental work. P.D. analyzed the data. P.D., H.M., and S.P.D. wrote the manuscript.
Funding Statement
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (2020‐05412, 2020‐00530, 590749‐24). P.D. acknowledges funding from the China Scholarship Council and the Tai Hung Fai Charitable Foundation. S.G.B. acknowledges funding from the Society for Laboratory Automation and Screening Graduate Education Fellowship Grant.
Conflicts of Interest
S.G.B. and H.M. have financial interest in ImageCyte Technologies, which is commercializing the nanowell‐in‐microwell plates. Some of the authors are inventors on patent applications own by the University of British Columbia.
Supporting information
Supporting File: smtd70493‐sup‐0001‐SuppMat.pdf.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1S. Kamiloglu , G. Sari , T. Ozdal , and E. Capanoglu , “Guidelines for Cell Viability Assays,” Food Frontiers 1 (2020): 332–349, 10.1002/fft 2.44. · doi ↗
- 2V. Vichai and K. Kirtikara , “Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening,” Nature Protocols 1 (2006): 1112–1116, 10.1038/nprot.2006.179.17406391 · doi ↗ · pubmed ↗
- 3A. Kummrow , M. Frankowski , N. Bock , C. Werner , T. Dziekan , and J. Neukammer , “Quantitative Assessment of Cell Viability Based on Flow Cytometry and Microscopy,” Cytometry Part A 83A (2013): 197–204, 10.1002/cyto.a.22213.23081720 · doi ↗ · pubmed ↗
- 4N. Atale , S. Gupta , U. C. S. Yadav , and V. Rani , “Cell‐Death Assessment By Fluorescent And Nonfluorescent Cytosolic And Nuclear Staining Techniques,” Journal of Microscopy 255 (2014): 7–19, 10.1111/jmi.12133.24831993 · doi ↗ · pubmed ↗
- 5J. Icha , M. Weber , J. C. Waters , and C. Norden , “Phototoxicity in Live Fluorescence Microscopy, and How to Avoid It,” Bio Essays 39 (2017): 1700003, 10.1002/bies.201700003.28749075 · doi ↗ · pubmed ↗
- 6L. Khalef , R. Lydia , K. Filicia , and B. Moussa , “Cell Viability and Cytotoxicity Assays: Biochemical Elements and Cellular Compartments,” Cell Biochemistry and Function 42 (2024): 4007, 10.1002/cbf.4007.38593323 · doi ↗ · pubmed ↗
- 7S. Park , V. Veluvolu , W. S. Martin , et al., “Label‐free, Non‐invasive, and Repeatable Cell Viability Bioassay Using Dynamic Full‐field Optical Coherence Microscopy and Supervised Machine Learning,” Biomedical Optics Express 13 (2022): 3187–3194, 10.1364/BOE.452471.35781969 PMC 9208588 · doi ↗ · pubmed ↗
- 8S. Li , Y. Li , J. Yao , et al., “Label‐Free Classification Of Dead And Live Colonic Adenocarcinoma Cells Based on 2D Light Scattering and Deep Learning Analysis,” Cytometry Part A 99 (2021): 1134–1142, 10.1002/cyto.a.24475.34145728 · doi ↗ · pubmed ↗