Expression Analysis of VEGF-Related Hub Genes and Pathways in Breast Cancer: A Comprehensive Bioinformatics Analysis
Mohadeseh Khoshandam, Mohammad Rahmanian, Mohammad Taghi Hedayati Goudarzi, Hossein Soltaninejad, Sadegh Babashah, Mahdiye Khoshandam

TL;DR
This study identifies key genes and pathways related to VEGF in breast cancer, suggesting potential targets for treatment.
Contribution
The study provides new insights into VEGF-related hub genes and pathways in breast cancer using comprehensive bioinformatics methods.
Findings
MMP9, MMP14, and VEGFA showed significant increases in expression in breast cancer.
Key pathways like receptor binding signaling and extracellular matrix regulation were enriched among differentially expressed genes.
Validation through microarray studies confirmed significant gene expression changes in breast cancer datasets.
Abstract
Breast cancer is the most common form of cancer among women worldwide, and the rates of both new cases and deaths have increased over the past two decades. The aim of the study was to identify and validate molecular pathways that could potentially be targeted for therapeutic interventions. The bioinformatics resource WebGestalt was used to determine the functional annotation of the Gene Ontology, as well as enrichment analysis of Reactome and KEGG pathways in 2023-2024. GeneMANIA, a server for assessing protein-gene interactions, co-localization, pathways, co-expression, and protein-domain similarity of target genes and their interacting genes, was evaluated via this web tool. GEO was also used to determine mRNA expression levels in BRCA individuals. R packages were used to screen for differentially expressed genes for both datasets. On the other hand, the open cancer resources GENT2…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
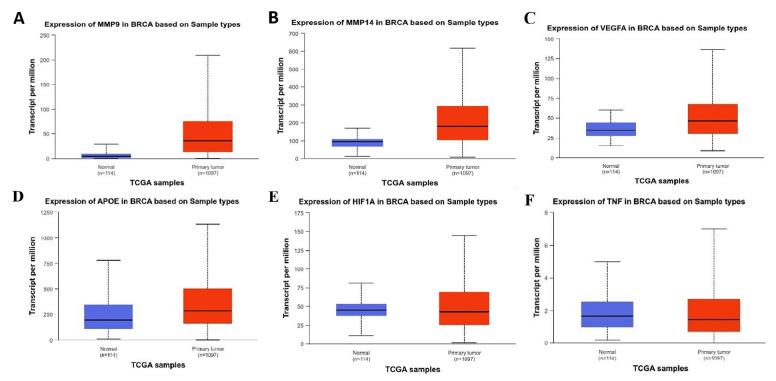 Figure 1
Figure 1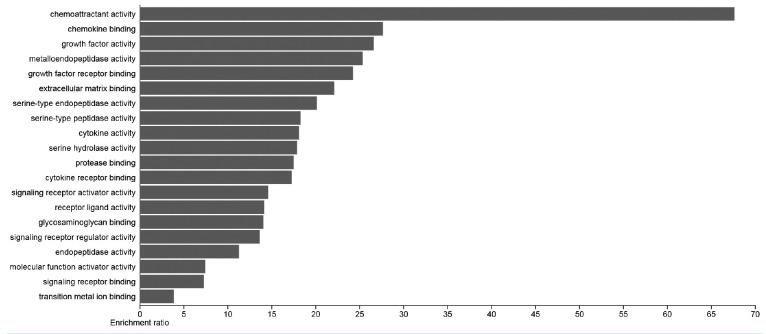 Figure 2
Figure 2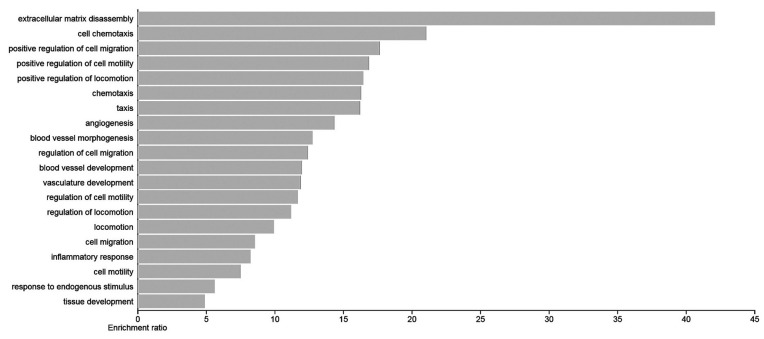 Figure 3
Figure 3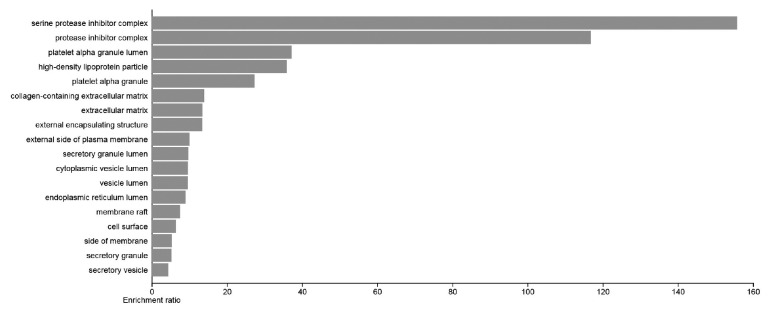 Figure 4
Figure 4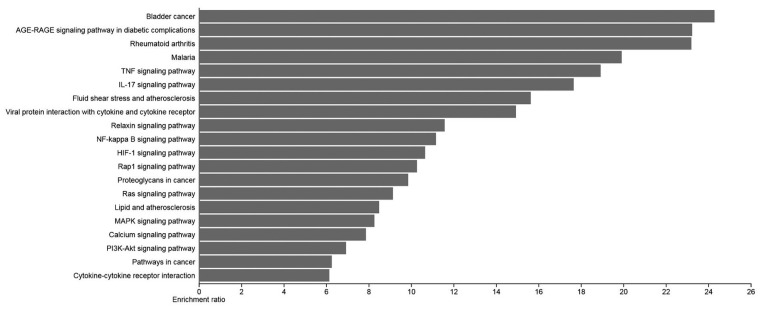 Figure 5
Figure 5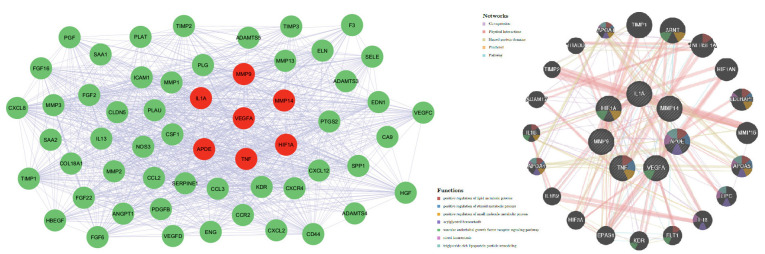 Figure 6
Figure 6| Genes | LogFC | P value | Adjusted P value |
|---|---|---|---|
|
| 2.7495 | 4.84E-41 | 1.02E-39 |
|
| 0.5549 | 8.57E-10 | 3.30E-09 |
|
| 0.6576 | 2.04E-06 | 5.87E-06 |
|
| 0.0806 | 0.4244 | 0.4992 |
|
| 0.05270 | 0.7505 | 0.8001 |
|
| 1.07134 | 3.51E-23 | 3.11E-22 |
|
| 0.4097 | 0.1063 | 0.1501 |
| Genes | ||||||
|---|---|---|---|---|---|---|
| LogFC | P value | Adjusted P value | Log FC | P value | Adjusted P value | |
|
| 1.41 | 4.12E-08 | 7.16E-07 | 2.45 | 7.83E-05 | 5.40E-04 |
|
| 0.28 | 0.00754 | 0.0237 | 0.418 | 0.0728 | 0.176 |
|
| 0.454 | 0.000067 | 0.000426 | 0.0497 | 0.8 | 0.8 |
|
| 0.264 | 0.0148 | 0.0412 | 0.255 | 0.269 | 0.46 |
|
| -0.0426 | 0.48 | 0.62 | 0.241 | 0.23 | 0.41 |
|
| 0.0373 | 0.633 | 0.745 | 0.0722 | 0.688 | 0.81 |
|
| 0.558 | 1.59E-06 | 0.000017 | 0.244 | 3.56E-02 | 1.01E-01 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBioinformatics and Genomic Networks · Machine Learning in Bioinformatics · Angiogenesis and VEGF in Cancer
What’s Known
- The literature highlights the central role of angiogenesis in tumor progression, metastasis, and therapeutic resistance.
What’s New
- Interaction network analysis identified MMP9, VEGFA, HIF1A, APOE, TNF, IL1A, MMP14, and TNF as central hub genes with extensive interactions across co-expression, physical interactions, and shared pathways. The results predict that the hub genes correlated with angiogenesis may serve as potential therapeutic targets or could be biomarkers for breast cancer.
Introduction
Breast cancer is a global health concern with a significant effect on public health. ^ 1 ^ There were about 2.26 million new cases of breast cancer in 2020, accounting for about 11.6% of all new cancer incidents and surpassing lung cancer as the most common cancer globally. ^ 2 ^ Among individuals diagnosed with early-stage breast cancer, approximately 30% of node-negative breast cancers and up to 70% of node-positive cancers can progress to metastatic disease, depending on various risk factors. ^ 3 ^ Understanding the genetic background of this malignancy is important for early detection, accurate diagnosis, and the development of specific treatments. ^ 4 ^
The formation of new blood vessels from preexisting ones in postnatal life, known as angiogenesis, plays a vital role in both normal bodily functions and pathological conditions. ^ 5 ^ The angiogenesis process is tightly regulated and involves the interaction of various pro- and anti-angiogenic factors. ^ 6 ^ When the balance between these factors is disrupted, it can result in either excessive or inhibited angiogenesis, which plays an important role in the development of different diseases. ^ 7 ^ Tumors cannot expand more than a certain volume unless they establish a blood supply to provide them with the necessary nutrients and oxygen for their growth. ^ 8 ^ Angiogenic growth factors, including epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and transforming growth factor (TGF), are crucial in facilitating angiogenesis and tumor development. ^ 9 ^ Among these factors, VEGF is pivotal in controlling the growth of endothelial cells, which are essential for the formation of new blood vessels. ^ 10 ^ VEGF is expressed in response to some soluble mediators, including growth factors and cytokines. By enhancing endothelial permeability, VEGF stimulates endothelial cell proliferation, survival, migration, cell-cell contact, and lumen formation. VEGF carries out its functions by binding to cell surface receptors, which are a group of membrane tyrosine kinase receptors. ^ 11 ^ By interacting with cell surface receptors, especially at the site of inflammation, it initiates intracellular signaling pathways and activates multiple genes that regulate essential cellular processes involved in angiogenesis. ^ 12 ^ Because tumor angiogenesis is crucial for tumor growth, studying the pathways and hub genes involved in it is seen as a promising approach to developing therapies that can hinder tumor growth and development. ^ 11 ^
Gene network formalism is the primary method used to uncover the role of genes that are overexpressed in cancer samples compared to normal ones. Innovations in in-silico techniques have streamlined research into gene function, disease, and precision medicine at the molecular level by employing an integrated approach to identify a group of genes linked to angiogenesis, migration, inflammation, and apoptosis that have a significant effect on the survival of breast cancer patients. ^ 13 ^ The objective of this study was to conduct a deep bioinformatics analysis of publicly available datasets to pinpoint hub genes and key pathways involved in angiogenesis and migration, particularly in their correlation with the inflammation pathway in breast cancer.
Materials and Methods
This study was conducted at the National Institute of Genetic Engineering and Biotechnology (NIGEB) in collaboration with Tarbiat Modares University of Iran, between 2023 and 2025.
Data Collection and Processing
Statistical analysis of differential gene expression was performed using the Limma-Voom pipeline. The empirical Bayes method implemented in the Limma package was used to moderate standard errors across genes. P values were adjusted for multiple testing using the Benjamini–Hochberg procedure. Genes with an adjusted P<0.05 and absolute log2 fold change greater than 1 were considered significantly differentially expressed.
This dataset initially contained 114 normal samples; however, one sample was excluded due to incomplete metadata, resulting in 113 normal samples and 1109 tumor samples used in the analysis.
WebGestalt (WEB-based GEne SeT AnaLysis Toolkit)
The bioinformatics resource called WebGestalt (https://www.webgestalt.org/) was employed to determine gene ontology (GO) functional annotation (GO terms, including Biological Process and Molecular Function). Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome pathways enrichment analysis were performed. An adjusted P< 0.05 was set to show a statistically significant difference.
GeneMANIA
GeneMANIA (https://genemania.org/) is a server for evaluating protein and genetic interactions, co-localization, pathways, co-expression, and domain-protein similarity of target genes. In this research, we assessed the relationship between matrix metalloproteinase-9 (MMP9), matrix metallopeptidase 14 (MMP14), Endogenous Vascular Endothelial Growth Factor-A (VEGFA), Apolipoprotein E (APOE), Hypoxia-Inducible Factor-1 Alpha (HIF1A), Tumor necrosis factor (TNF), Interleukin-1 alpha precursor (IL1A), and their interactive genes via this web tool.
Verification of Expression
The GEO database (https://www.ncbi.nlm.nih.gov/geo/) was used to determine the mRNA expression levels in BRCA (BReast CAncer gene) subjects. Two datasets of microarray, namely GSE37751 and GSE42568, were downloaded. R packages (affy and Limma) were used to screen differentially expressed genes for both datasets. P value less than 0.05 and logFC higher and lower than 0 were the filter conditions to explore DEGs between BRCA and normal specimens. On the other hand, the open cancer sources GENT2 (http://gent2.appex.kr/gent2/), TNMplot (https://tnmplot.com/), UCSCXena (https://xenabrowser.net/), ENCORI platform (https://rnasysu.com/encori/index.php), BioXpress (https://hivelab.biochemistry.gwu.edu/bioxpress), OncoDB (https://oncodb.org/), OncoMX (https://www.oncomx.org/), and GEPIA2 (http://gepia2.cancer-pku.cn/#index) were employed to assay differential expression of mRNAs in patients with BRCA.
Results
We performed a comprehensive analysis of angiogenic hub genes (MMP9, MMP14, VEGFA, APOE, HIF1A, TNF, IL1A) to investigate their expression patterns and biological relevance in breast cancer, focusing on their roles in inflammation and cell migration
Differential Gene Expression
Using RNA-seq data from TCGA and subsequent normalization through Limma and Voom packages, we identified significant changes in the expression of angiogenic hub genes. Among the analyzed genes, MMP9 exhibited the highest positive fold change with a logFC of 2.75 (figure 1A and table 1). Similarly, MMP14 showed substantial upregulation with a logFC of 1.07 (figure 1B and table 1). Other notable upregulated genes included VEGFA with a logFC of 0.55 (figure 1C and table 1), and APOE with a logFC of 0.66 (figure 1D and table 1).
The expression of MMP9 in BRCA and normal tissue is shown in panel A. The expression of MMP14 in BRCA and normal tissues is shown in panel B. The expression of VEGFA in BRCA and normal tissues is shown in panel C. The expression of APOE in BRCA and normal tissues is shown in panel D. The expression of APOE in BRCA and normal tissues is shown in panel E. The expression of HIF1A in BRCA and normal tissue is shown in panel F. RCA: Breast invasive carcinoma; MMP9: Matrix metalloproteinase 9; MMP14: Matrix metalloproteinase 14; VEGFA: Vascular endothelial growth factor A; APOE: Apolipoprotein E; TCGA: The cancer genome atlas; HIF1A: Hypoxia-inducible factor 1 Alpha; TNF: Tumor necrosis factor; BRCA: Breast invasive carcinoma. RCA: Breast invasive carcinoma; MMP9: Matrix metalloproteinase 9; MMP14: Matrix metalloproteinase 14; VEGFA: Vascular endothelial growth factor A; APOE: Apolipoprotein E; TCGA: The cancer genome atlas; HIF1A: Hypoxia-inducible factor 1 Alpha; TNF: Tumor necrosis factor; BRCA: Breast invasive carcinoma
In contrast, HIF1A displayed minimal expression changes (LogFC=0.08) with non-significant P values (figure 1E and table 1), indicating a limited role under the conditions analyzed. Similarly, TNF exhibited negligible changes in expression (figure 1F and table 1), suggesting no significant involvement in this dataset. Finally, IL1A demonstrated a slight increase in expression (LogFC=0.41), but this change was not statistically significant.
Validation Through Microarray Studies
The validation of gene expression changes through microarray studies in datasets GSE37751 and GSE42568 provided consistent and significant results for several genes. MMP9 showed strong upregulation in both datasets, with a LogFC of 1.41 in GSE37751 and 2.45 in GSE42568 (P=7.83E^-05^, adjusted P=5.40E^-04^), confirming its critical role in breast cancer progression. Similarly, MMP14 demonstrated significant upregulation in GSE37751, while in GSE42568, its expression was moderately elevated, providing partial validation of its role.
VEGFA showed moderate upregulation in GSE37751 and higher expression in GSE42568, suggesting its involvement in angiogenesis, although its statistical significance was weaker in GSE42568. APOE displayed slight upregulation in GSE37751, indicating its relevance to lipid metabolism in breast cancer, but its expression in GSE42568 was not significant, reflecting variability across datasets.
On the other hand, HIF1A showed limited but significant upregulation in GSE37751, while its expression in GSE42568 was not statistically significant, suggesting a minor or dataset-specific role. TNF exhibited minimal expression changes in both datasets, with a slight downregulation in GSE37751 and marginal upregulation in GSE42568, confirming no significant involvement. Similarly, IL1A showed negligible changes in expression, with LogFC values of 0.0373 in GSE37751 and 0.0722 in GSE42568, indicating no significant role in breast cancer (table 2).
Functional Enrichment Analysis
GO analysis revealed significant molecular functions and biological processes enriched among the DEGs.
Molecular Functions
The top enriched activities include “chemoattractant activity” and “chemokine binding”, highlighting the involvement of these genes in immune cell recruitment and inflammatory responses (figure 2). Among the top pathways, signaling receptor binding, signaling receptor activator activity, signaling receptor regulator activity, receptor ligand activity, and molecular function activator activity were identified as superior based on FDR and P value.
Bar plot of the top 20 Molecular Function (MF) terms. Each row shows an enrichment function. The bar graph length shows the enrichment ratio. The enrichment ratio is determined by dividing the number of input genes by the number of background genes.
Biological Processes
The most enriched pathways include “extracellular matrix disassembly” and “cell chemotaxis”, indicating the involvement of these genes in cell migration and metastatic processes (figure 3). Among the top pathways, regulation of cell migration and cell motility, locomotion, regulation of locomotion, and cell migration were identified as superior based on FDR and P value.
The bar plot of the top 20 Biological Process (BP) terms is shown here. Each row shows an enrichment function. The bar graph length shows the enrichment ratio. The enrichment ratio is determined by dividing the number of input genes by the number of background genes.
Cellular Components
Genes were predominantly localized in “serine protease inhibitor complexes” and “collagen-containing extracellular matrix”, underscoring their roles in structural tissue remodeling (figure 4). Among the top pathways, extracellular matrix, external encapsulating structure, collagen-containing extracellular matrix, platelet alpha granule lumen, and cell surface were identified as superior based on FDR and P value.
The bar plot of the top 20 Cellular Component (CC) terms is shown here. Each row represents an enrichment function. Each row shows an enrichment function. The bar graph length shows the enrichment ratio. The enrichment ratio is calculated by dividing the count of input genes by the count of background genes.
KEGG Pathways
The genes were enriched in pathways, including “Bladder cancer”, “AGE-RAGE signaling in diabetic complications”, and “TNF signaling”, reflecting their diverse functional implications in cancer progression and angiogenesis (figure 5). Among the top pathways, the AGE-RAGE signaling pathway in diabetic complications, Rheumatoid arthritis, TNF signaling pathway, Fluid shear stress and atherosclerosis, and Pathways in cancer were identified as superior based on FDR and P value.
The bar chart of enrichment ratios of the top 20 significantly enriched KEGG pathways is shown in this figure. The enrichment ratio represents the proportion of observed genes to the expected genes within the gene list for the KEGG category.
Network Analysis
Interaction network analysis identified MMP9, VEGFA, HIF1A, APOE, TNF, IL1A, MMP14, and TNF as central hub genes with extensive interactions across co-expression, physical interactions, and shared pathways. VEGFA is primarily involved in the vascular endothelial growth factor receptor signaling pathway. HIF1A contributes to the positive regulation of lipid metabolic processes and small-molecule metabolic processes. APOE plays a role in the positive regulation of lipid metabolic processes and contributes to acylglycerol homeostasis. In addition, APOE is crucial in the particle remodeling of triglyceride-rich lipoprotein. TNF plays a role in regulating lipid metabolism, particularly in inflammatory responses, which can influence lipid storage and breakdown. Additionally, it is involved in regulating steroid metabolism, often in the context of stress and inflammatory signaling also participates in regulating various small-molecule metabolic pathways, such as glucose and amino acid metabolism, often during immune responses. These genes are pivotal in regulating angiogenesis, cell migration, and inflammatory responses in breast cancer (figure 6).
The MMP-9, MMP-14, VEGFA, APOE, HIF1A, TNF, and IL1A genes’ interaction network and related functions were obtained by GenMANIA analysis.
Cross-Database Validation
Validation of expression trends was performed using various databases, including UALCAN, GENT2, TNMPlot, and UCSC Xena. Genes such as MMP9, MMP14, VEGF, APOE, HIF1A, TNF, and IL1A were consistently upregulated, while prostaglandin-endoperoxide synthase 2 (PTGS2), B-cell leukemia/lymphoma 2 protein (BCL2), and interleukin 6 (IL6) were downregulated among the datasets that were not included in our study.
Additional Pathway Analysis
Further exploration of pathways emphasized the critical roles of angiogenic hub genes in breast cancer progression. The overlap between bladder cancer and breast cancer highlighted shared mechanisms in ECM remodeling and immune modulation, involving components such as MMP9, VEGFA, and TNF. The AGE-RAGE signaling pathway was relevant to chronic inflammation and angiogenesis, driven by VEGFA and TNF. The interleukin 17 (IL-17) signaling pathway involves genes such as IL1A, IL1B, and TNF, amplifying inflammatory responses and promoting tumor progression. The fluid shear stress and atherosclerosis pathway highlighted vascular adaptations in the tumor microenvironment, mediated by VEGFA and TNF. The HIF-1 signaling pathway played a key role in hypoxia responses through HIF1A, VEGFA, and MMP9.
Discussion
Interaction network analysis in our study identified MMP9, VEGFA, HIF1A, APOE, TNF, IL1A, MMP14, and TNF genes as core genes with extensive interactions in co-expression, physical interactions, and shared pathways. The results predicted that core genes related to angiogenesis may serve as potential therapeutic targets or biomarkers for breast cancer.
The breast cancer management continues to pose significant challenges. Understanding the protein families and pathways that contribute to the development and progression of cancer is a vital step in the development of targeted treatments.
VEGF-A is a well-known mediator of tumor angiogenesis in many solid tumors and has been used as an angiogenesis inhibitor in breast cancer. ^ 14 ^ Additionally, high levels of MMP-14 and MMP-9 expression are associated with increased TNF expression in the tumor stroma and different survival rates compared to each other. ^ 15
- 17 ^ Various studies have shown that increased levels of inflammatory factors, such as TNF, contribute to the promotion of breast cancer stem-like cells. ^ 18 ^ It was also demonstrated that TNF enhances breast cancer stem-like cells by upregulating TAZ expression through the non-canonical signaling (NCS) of the NF-κB pathway. Understanding the signaling cascade initiated by VEGF and the subsequent effect of the HIF gene on the genome of cancer cells may lead to breakthroughs in personalized treatment of solid cancers, including breast cancer. ^ 19 , 20 ^ APOE has been regarded as a potential biomarker that predicts the prognosis of cancer and has been associated with immune infiltration. ^ 21 ^ IL-1A produced by tumor cells influenced breast cancer progression and metastasis in mice by modulating the tumor immune microenvironment. ^ 22 ^
In this study, we investigated the relationship between VEGF, MMP-9, MMP-14, APOE, HIF1A, TNF, and IL-1A and breast cancer, and their upregulation in breast cancer cells was observed. Pathways involved in 1109 breast cancer tumor samples were compared with 113 normal samples, and the results were confirmed using online databases.
We also explored the MAPK signaling pathway, TNF signaling pathway, Rap1 signaling pathway, Ras signaling pathway, IL-17 signaling pathway, PI3K-Akt signaling pathway, lipid and atherosclerosis pathway, NF-kappa B signaling pathway, and HIF-1 signaling pathway.
MAPK signaling is associated with various types of cancer in humans, and its activation is influenced by a variety of external and internal signals and proteins. ^ 23 ^ Consequently, concentrating on these aspects of the signaling pathway often results in adverse effects, activation of alternative mechanisms, and reduced efficacy of the drug. This highlights the importance of a more comprehensive therapeutic approach. To mitigate these detrimental outcomes, it is advisable to explore alternative strategies that center on RAF modulators. ^ 24 ^ The spatiotemporal attributes of the MAPK pathway components could provide a novel perspective.
Structure, upstream activators, and downstream factors play a critical role in Erk (MAPK) kinase, a vital component of the Ras/Raf/MAPK pathway, which is responsible for regulating cellular metabolism in cancer cells. ^ 25 ^
The Ras/Raf/MAPK pathway can be activated through two mechanisms: a ligand-dependent mechanism, in which ligands such as hormones, cytokines, or growth factors bind physically to the receptors; and a ligand-independent mechanism, which can be initiated by physical stressors such as injury, radiation, or changes in osmotic pressure. This pathway plays a role in the growth of cancerous cells in humans. ^ 25 , 26 ^
Aside from mutations in the Ras gene, disruptions in components that regulate Ras can also affect Raf activation. During various biological processes, different growth factors, such as platelet-derived growth factor BB (PDGF-BB), EGF, TGF-α, and VEGF, can stimulate the canonical Ras-Raf-MEK-Erk pathway. ^ 27 ^ Further studies on Raf activation will help reveal the important molecular mechanisms driving cell proliferation, and metastasis and survival in cancer, especially through the roles of small Ras-GTPases and the EGF receptor (EGFR). As such, extensive research is currently underway to identify Raf kinase as a promising target for the development of anticancer drugs. ^ 28 , 29 ^
A small proportion of resistant melanomas depends on compensatory PI3K (phosphatidylinositol 3-kinase) and Akt (protein kinase B) signaling. ^ 30 ^ Early clinical trials involving PI3K inhibitors alongside vemurafenib demonstrated promising results. ^ 31 ^ EGFR has shown efficacy in BRAFV600E-mutated CRCs, by activating the PI3K/AKT pathway serving as a pathway for both acquired and innate resistance to BRAF inhibitors. ^ 32 ^
The resistance to chemotherapy caused by hypoxia poses a significant challenge in treating solid tumors. HIF1α, a gene that helps the body adapt to low oxygen levels, was found to have a profound impact on the efficacy of chemotherapy for various types of cancer and specific treatment plans. ^ 33 ^ The results of multivariate and univariate logistic regression analysis indicated that high levels of HIF1α were associated with lower pathologic complete response rates and a poorer prognosis. ^ 34 ^ The analysis of the geospatial dataset also revealed a negative correlation between HIF1α expression and the response to neoadjuvant drugs in breast cancer patients. ^ 35 ^
KEGG enrichment of DEGs of different HIF1α expression groups in the TCGA database revealed that HIF1α is involved in the IL-17 signaling pathway. ^ 36 ^ Additionally, correlation analysis showed a positive relationship between HIF1α and the IL-17 pathway. ^ 37 ^ Laboratory tests found that the HIF1α/IL-17 pathway affects the sensitivity of breast cancer cells to paclitaxel. ^ 37 ^ Furthermore, correlation analysis suggested that high expression of HIF1α/IL-17A/ C-X-C motif chemokine ligand 10 (CXCL10) genes is positively associated with neutrophil infiltration in breast cancer. ^ 37 ^
NF-κB serves as a key mediator of inflammation and cancer, with known implications in breast cancer and endocrine disorders. NF-κB is regulated by the canonical signaling (CS) pathway (activated by lipopolysaccharide [LPS], Reactive oxygen species [ROS], TNF-α, and IL-1) and the NCS pathways (triggered by inflammatory stimuli of inhibitor of nuclear factor kappa-B kinase subunit alpha [IKK-α]). ^ 38 ^ Dysregulation of NF-κB can lead to cancer, and its activation is observed in different cancers, such as breast cancer.
The NF-κB role in tumor development is intricate due to the multiple functions of the tumor microenvironment. ^ 39 ^ Upregulation of NF-κB in tumor cells can induce inflammation in the tumor microenvironment, leading to increased cytokine gene expression and cytokine release. ^ 40 ^ This results in activating the classical NF-κB signaling pathway and subsequent apoptosis of transformed cells. ^ 41 ^ Hence, NF-κB may serve as a potential marker and therapeutic target for breast cancer prediction and treatment. Despite the cytotoxic effects of drugs in tumor eradication, they are limited by potential side effects.
Although there is an imbalance between the number of tumors (n=1109) and normal (n=113) samples in the TCGA dataset, the Limma-Voom framework is well-suited for such scenarios. It uses precision weights to model the mean-variance relationship in RNA-seq data, which improves the reliability of differential expression estimates even when group sizes are unequal. Furthermore, the empirical Bayes moderation implemented in Limma stabilizes variance estimates across genes, mitigating the effects of sample size disparity. Therefore, while the imbalance is acknowledged as a potential limitation, the statistical framework employed here helps minimize its practical impact on the results.
Conclusion
The development of breast cancer is influenced by a complex interaction between environmental and genetic factors. Researchers found that specific genes, referred to as regulatory hub genes, could serve as potential targets for drug-based therapies. This discovery was made by analyzing extensive datasets of microarray and RNA sequencing data that are linked to pathways such as angiogenesis, migration, and inflammation. These identified hub genes are part of the protein family and have the potential to play a role in the progression of breast cancer. By recognizing these genes as potential targets for the treatment of breast cancer, new and more advanced therapeutic strategies could be developed.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Khoshandam M Soheili ZS Hosseinkhani S Samiee S Latifi-Navid H Ahmadieh Hetal In vivo inhibition of angiogenesis by hts FLT 01/Mi RGD nano complex Transl Oncol 202556102400[ PMC Free Article ]10.1016/j.tranon.2025.10240040306151 PMC 12434996 · doi ↗ · pubmed ↗
- 2Wu B Li Y Shi B Zhang X Lai Y Cui Fetal Temporal trends of breast cancer burden in the Western Pacific Region from 1990 to 2044: Implications from the Global Burden of Disease Study 2019 J Adv Res 20245918999[ PMC Free Article ]10.1016/j.jare.2023.07.00337422280 PMC 11082062 · doi ↗ · pubmed ↗
- 3Cherny NI Paluch-Shimon S Berner-Wygoda Y Palliative care: needs of advanced breast cancer patients Breast Cancer (Dove Med Press). 20181023143[ PMC Free Article ]10.2147/BCTT.S 16046230584354 PMC 6284851 · doi ↗ · pubmed ↗
- 4De Berardinis RJ Tumor Microenvironment, Metabolism, and Immunotherapy N Engl J Med 20203828697110.1056/NEJ Mcibr 191489032101671 · doi ↗ · pubmed ↗
- 5Manning D Rivera EJ Santana LF The life cycle of a capillary: Mechanisms of angiogenesis and rarefaction in microvascular physiology and pathologies Vascul Pharmacol 2024156107393[ PMC Free Article ]10.1016/j.vph.2024.10739338857638 PMC 12051481 · doi ↗ · pubmed ↗
- 6Shaw P Dwivedi SKD Bhattacharya R Mukherjee P Rao G VEGF signaling: Role in angiogenesis and beyond Biochim Biophys Acta Rev Cancer 2024187918907910.1016/j.bbcan.2024.18907938280470 PMC 12927493 · doi ↗ · pubmed ↗
- 7Akbarian M Bertassoni LE Tayebi L Biological aspects in controlling angiogenesis: current progress Cell Mol Life Sci 202279349[ PMC Free Article ]10.1007/s 00018-022-04348-535672585 PMC 10171722 · doi ↗ · pubmed ↗
- 8Sriharikrishnaa S Suresh PS Prasada KS An introduction to fundamentals of cancer biology. In: Optical Polarimetric Modalities for Biomedical Research 2023 Cham Springer 3073010.1007/978-3-031-31852-8_11 · doi ↗