Correction to “Spatial Reorganization of Chromatin Architecture Shapes the Expression Phenotype of Therapy‐Induced Senescent Cells”

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
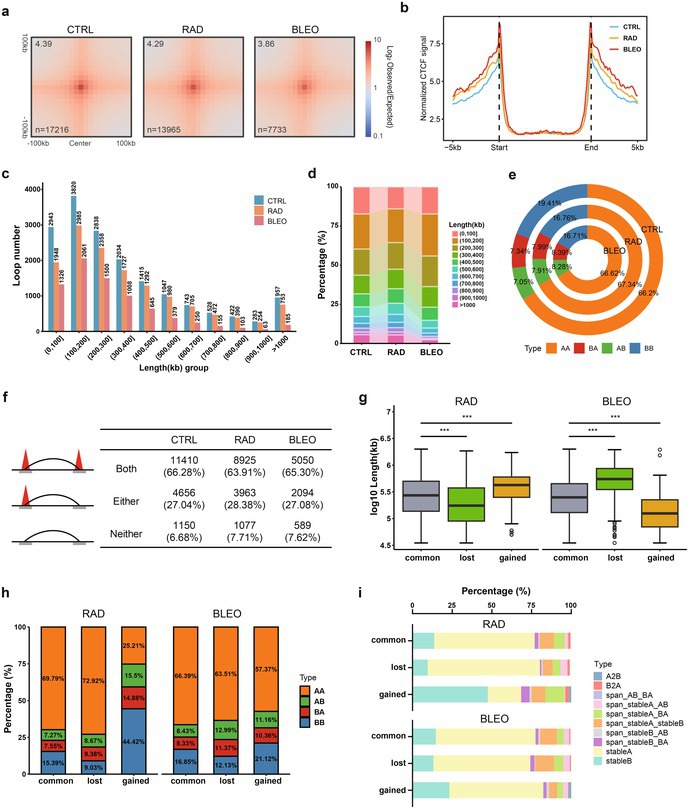 Figure 1
Figure 1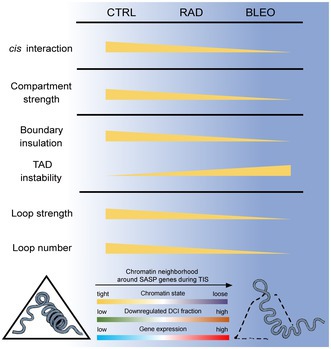 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTelomeres, Telomerase, and Senescence · Genomics and Chromatin Dynamics · Pluripotent Stem Cells Research
Zhang, G., W. Zhang, C. Wang, et al. 2026. “Spatial Reorganization of Chromatin Architecture Shapes the Expression Phenotype of Therapy‐Induced Senescent Cells.” Aging Cell 25, no. 1: e70366. https://doi.org/10.1111/acel.70366.
In the published version of the above article, we identified several minor errors, including small wording and formatting issues. These have now been corrected as detailed below. This correction does not affect the results or conclusions of the article.
Main text:
(1) In section 2.3, third paragraph, in the following sentence:
‘Across short‐and long‐range interactions within both A and B compartments, RAD showed significantly weaker compartmentalization than CTRL, whereas BLEO exhibited significantly stronger compartmentalization in short‐range A and long‐range B compared with CTRL (Figure 3b and Table S3).’ “and BLEO” should be added after “RAD” and the term “CTRL” should be corrected to “RAD”.
The corrected sentence should now read as follows:
Across short‐and long‐range interactions within both A and B compartments, RAD and BLEO showed significantly weaker compartmentalization than CTRL, whereas BLEO exhibited significantly stronger compartmentalization in short‐range A and long‐range B compared with RAD (Figure 3b and Table S3).
(2) In section 2.5 third paragraph, in the following sentence:
‘Approximately 66% of loops in all 3 conditions fell into the AA type, which was also the predominant configuration across different loop lengths (Figures 5d and S7b)’.
‘(Figures 5d and S7b)’ should be corrected to ‘(Figures 5e and S7b)’.
(3) In section 2.8, page 12, third paragraph, the phrase “and Figure S11d” should be added after “Figure 6d”.
(4) In the Methods section, subsection 4.9, first paragraph, in the following sentence,
‘For the analysis of OIS and RS Hi‐C data, we employed the same reference genome and processing pipeline as used in this study.’ the phrase “OIS and RS” should be corrected to “OIS, RS and deep RS” and the corrected sentence should now read as
**‘**For the analysis of OIS, RS and deep RS Hi‐C data, we employed the same reference genome and processing pipeline as used in this study’.
(5) In section 4.12, line 2, the term “fdr 0.1” should be corrected to “FDR < 0.1”.
(6) In the Data Availability Statement, the term “CUT&Tag” should be corrected to “CUT&RUN” and the sentence “H3K9me3 ChIP‐seq data referred to former datasets in GSE163105.” should be removed, as this dataset was not used in the study.
(7) In Figure caption 2, lines 1–2, the chromosome number in Figure 2a and b should be “4” instead of “11” and in Figure 2i, the phrase “change of intra‐ and inter‐ interactions” should be corrected to “changes of interactions.”
(8) In Figure 5a, line 2, the term “The top right corner” should be corrected to “The top left corner”.
(9) In Figure 6a, the term “reorganization” should be corrected to “rearrangement events”
(10) In Figure 6f, the sentence “Euclidean distance between the location of NRG1 and other locations in the TAD represented by the blue line segment.” should be corrected to “Boxplot of comparison of Euclidean distance between the location of NRG1 in red and other locations represented by the blue line segment in e.”
(11) Figure 5e appeared to be distorted, and a legend title was inadvertently added to Figure 5i. The corrected version of Figure 5 was shown below
(12) The incorrect version of Figure 7 was updated in the published version, the trend shown in the figure did not match the results described in the article. The corrected version of Figure 7 was shown below
(13) We missed to include the reference citations to bioinformatics tools and the references in reference list. The citations and the missed out references were listed below:
- In Methods section, subsection 4.3, in second paragraph, (Kim et al. 2015) should be cited after Hisat2 (version 2.2.1), (Anders et al. 2015) after HTSeq (version 2.0.2), (Pertea et al. 2015) after StringTie (version 2.2.1), (Love et al. 2014) after DESeq2 (version 1.34.0) and (Dennis Jr. et al. 2003) after DAVID.
- In subsection 4.7, (Langmead and Salzberg 2012) should be cited after Bowtie2 (version 2.3.5.1), (Li et al. 2009) after SAMtools (version 1.3.1), (Feng et al. 2012) should be cited at the end of the text ‘MACS2 was applied to each merged bam file to call peaks’, (Ramírez et al. 2014) after deepTools (version 3.5.1).
- In subsection 4.9, (Servant et al. 2015) should be cited after HiC‐Pro (version 3.1.0) pipeline, (Ramírez et al. 2018) after HiCExplorer (version 3.7.2).
- In subsection 4.10, (van der Weide et al. 2021) should be cited after ‘GENOVA’, (Magnitov et al. 2022) after ‘Pentad’.
- In subsection 4.11, (Flyamer et al. 2020) should be cited after ‘coolpuppy (version 1.1.0)’, (Cao et al. 2022) after ‘cLoops2 (version 0.0.3)’.
- In subsection 4.13, (Stansfield et al. 2019) should be cited after ‘multiHiCcompare (version 1.20.0)’.
- **In subsection 4.14, (**Yang et al. 2017 **) should be cited after ‘**HiCRep’.
We apologize for these errors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Anders, S. , P. T. Pyl , and W. Huber . 2015. “HT Seq—A Python Framework to Work With High‐Throughput Sequencing Data.” Bioinformatics (Oxford, England) 31: 166–169. 10.1093/bioinformatics/btu 638.25260700 PMC 4287950 · doi ↗ · pubmed ↗
- 2Cao, Y. , S. Liu , G. Ren , Q. Tang , and K. Zhao . 2022. “c Loops 2: A Full‐Stack Comprehensive Analytical Tool for Chromatin Interactions.” Nucleic Acids Research 50: 57–71. 10.1093/nar/gkab 1233.34928392 PMC 8754654 · doi ↗ · pubmed ↗
- 3Dennis, G., Jr. , B. T. Sherman , D. A. Hosack , et al. 2003. “DAVID: Database for Annotation, Visualization, and Integrated Discovery.” Genome Biology 4: P 3.12734009 · pubmed ↗
- 4Feng, J. , T. Liu , B. Qin , Y. Zhang , and X. S. Liu . 2012. “Identifying Ch IP‐seq Enrichment Using MACS.” Nature Protocols 7: 1728–1740. 10.1038/nprot.2012.101.22936215 PMC 3868217 · doi ↗ · pubmed ↗
- 5Flyamer, I. M. , R. S. Illingworth , and W. A. Bickmore . 2020. “Coolpup.py: Versatile Pile‐Up Analysis of Hi‐C Data.” Bioinformatics (Oxford, England) 36: 2980–2985. 10.1093/bioinformatics/btaa 073.32003791 PMC 7214034 · doi ↗ · pubmed ↗
- 6Kim, D. , B. Langmead , and S. L. Salzberg . 2015. “HISAT: A Fast Spliced Aligner With Low Memory Requirements.” Nature Methods 12: 357–360. 10.1038/nmeth.3317.25751142 PMC 4655817 · doi ↗ · pubmed ↗
- 7Langmead, B. , and S. L. Salzberg . 2012. “Fast Gapped‐Read Alignment with Bowtie 2.” Nature Methods 9: 357–359. 10.1038/nmeth.1923.22388286 PMC 3322381 · doi ↗ · pubmed ↗
- 8Li, H. , B. Handsaker , A. Wysoker , et al. 2009. “The Sequence Alignment/Map Format and SA Mtools.” Bioinformatics (Oxford, England) 25: 2078–2079. 10.1093/bioinformatics/btp 352.19505943 PMC 2723002 · doi ↗ · pubmed ↗