Blood PCSK9 Impacts Alzheimer's Disease Risk in an APOE Genotype‐Dependent Manner: A Prospective Cohort Study
Qiushan Tao, Ting Fang Alvin Ang, Jinghan Huang, Indira Swetha Itchapurapu, Jesse Mez, Michael Alosco, Rhoda Au, Lindsay A. Farrer, Xiaoling Zhang, Wei Qiao Qiu

TL;DR
High blood levels of PCSK9 may protect against Alzheimer's disease, but only in people without the APOE ε4 gene variant.
Contribution
This study shows that PCSK9's effect on Alzheimer's risk depends on APOE genotype, suggesting personalized treatment approaches.
Findings
High plasma PCSK9 levels are linked to lower Alzheimer's risk in APOE ε4 noncarriers.
PCSK9 genotypes associated with low levels increase Alzheimer's risk only in APOE ε4 noncarriers.
Findings were validated using the ADNI study and CSF biomarkers.
Abstract
Apolipoprotein E (APOE) and proprotein convertase subtilisin/kexin type 9 (PCSK9) are both lipid proteins and related to immunity/inflammation. We hypothesized that PCSK9 impacts on Alzheimer's disease (AD) risk in an APOE genotype dependent manner. We used the Framingham Heart Study (FHS) Offspring cohort (Gen 2), with data on plasma PCSK9 protein concentration, as the baseline exposure for 1,704 study subjects. Using Cox regression models, the outcomes were incidents of AD or all‐cause dementia. Using another FHS dataset with 3,048 individuals with genetic data, we examined the association between PCSK9 genotypes and the incidence of AD/dementia, stratifying the analysis based on APOE ε4 status. The Alzheimer's Disease Neuroimaging Initiative (ADNI) study was used to validate some of the main findings. Higher plasma PCSK9 protein levels were associated with a lower risk of AD (HR…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
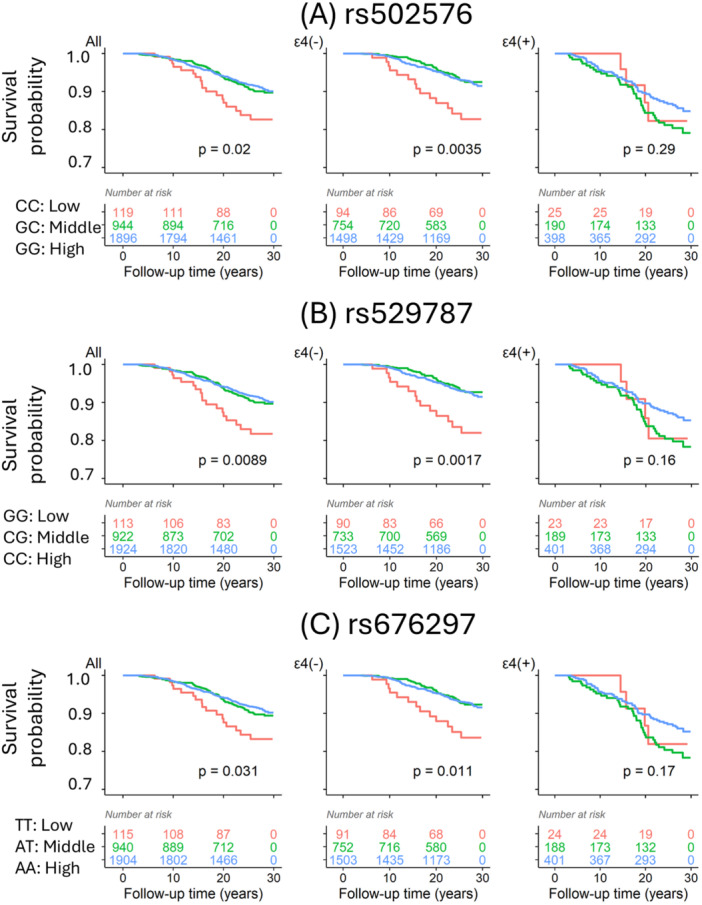 Figure 1
Figure 1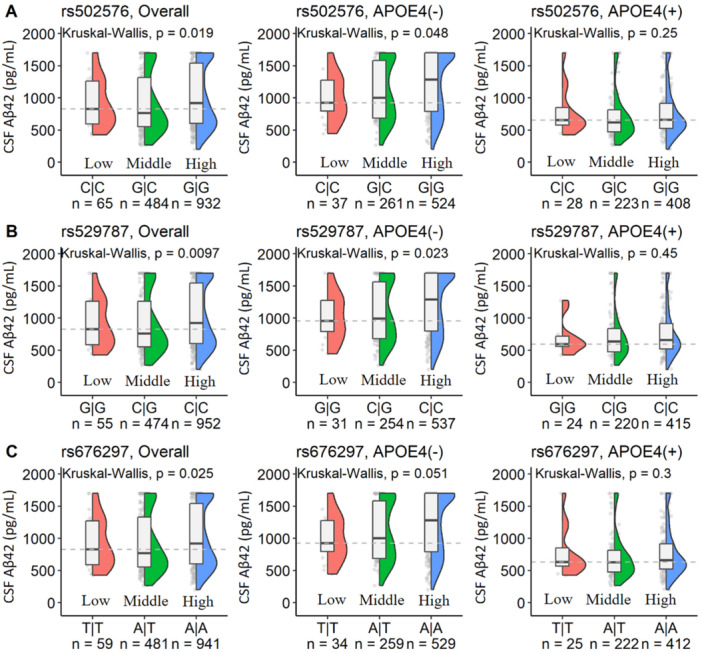 Figure 2
Figure 2| Models | Exposure | Alzheimer's disease | All‐cause dementia | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| Events | HR [95% CI] |
|
| Events | HR [95% CI] |
| ||
|
| |||||||||
| PCSK9 | 1655 | 136 | 0.85 [0.73, 0.99] | 0.043 | 1704 | 185 | 0.88 [0.77, 1.01] | 0.080 | |
|
| |||||||||
| PCSK9 | 1634 | 135 | 0.94 [0.79, 1.12] | 0.471 | 1682 | 183 | 0.97 [0.84, 1.13] | 0.707 | |
|
| |||||||||
| PCSK9 | 1634 | 135 | 0.80 [0.65, 0.99] | 0.039 | 1682 | 183 | 0.85 [0.71, 1.01] | 0.064 | |
| Interaction | 1634 | 135 | 1.59 [1.12, 2.27] | 0.010 | 1682 | 183 | 1.54 [1.13, 2.09] | 0.006 | |
|
| |||||||||
| PCSK9 | 1585 | 127 | 0.71 [0.57, 0.90] | 0.004 | 1629 | 171 | 0.81 [0.66, 0.98] | 0.033 | |
| Interaction | 1585 | 127 | 1.80 [1.24, 2.62] | 0.002 | 1629 | 171 | 1.65 [1.20, 2.28] | 0.002 | |
| Strata | Models | Alzheimer's disease | All‐cause dementia | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| Events | HR [95CI] |
|
| Events | HR [95CI] |
| ||
|
| |||||||||
|
| 1290 | 93 | 0.78 [0.63, 0.96] | 0.021 | 1325 | 128 | 0.83 [0.69, 0.99] | 0.040 | |
|
| 1250 | 87 | 0.74 [0.58, 0.94] | 0.013 | 1281 | 118 | 0.84 [0.69, 1.03] | 0.100 | |
|
| |||||||||
|
| 344 | 42 | 1.29 [0.97, 1.73] | 0.084 | 357 | 55 | 1.33 [1.03, 1.72] | 0.030 | |
|
| 335 | 40 | 1.22 [0.85, 1.74] | 0.280 | 348 | 53 | 1.25 [0.92, 1.70] | 0.159 | |
| Strata | SNP | Genotype | Alzheimer's disease | All‐cause dementia | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| Events | HR [95CI] |
|
| Events | HR [95CI] |
| |||
|
|
| CC: Low | 94 | 14 | ref | — | 96 | 16 | ||
| GC: Middle | 740 | 46 | 0.28 [0.15, 0.52] | < 0.001 | 762 | 68 | 0.40 [0.23, 0.69] | 0.001 | ||
| GG: High | 1482 | 99 | 0.30 [0.17, 0.53] | < 0.001 | 1523 | 140 | 0.41 [0.24, 0.70] | < 0.001 | ||
|
| GG: Low | 90 | 14 | ref | — | 92 | 16 | ref | — | |
| CG: Middle | 733 | 44 | 0.26 [0.14, 0.49] | < 0.001 | 755 | 66 | 0.38 [0.22, 0.66] | < 0.001 | ||
| CC: High | 1523 | 101 | 0.29 [0.17, 0.51] | < 0.001 | 1566 | 144 | 0.40 [0.24, 0.67] | < 0.001 | ||
|
| TT: Low | 91 | 13 | ref | — | 93 | 15 | ref | — | |
| AT: Middle | 739 | 47 | 0.30 [0.16, 0.56] | < 0.001 | 758 | 66 | 0.41 [0.23, 0.72] | 0.002 | ||
| AA: High | 1486 | 99 | 0.32 [0.18, 0.57] | < 0.001 | 1530 | 143 | 0.44 [0.25, 0.75] | 0.003 | ||
|
|
| CC: Low | 25 | 4 | ref | — | 26 | 5 | ref | — |
| GC: Middle | 186 | 33 | 1.62 [0.57, 4.63] | 0.37 | 195 | 42 | 1.52 [0.60, 3.89] | 0.38 | ||
| GG: High | 395 | 51 | 1.36 [0.48, 3.84] | 0.56 | 406 | 62 | 1.26 [0.50, 3.20] | 0.62 | ||
|
| GG: Low | 23 | 4 | ref | — | 24 | 5 | ref | — | |
| CG: Middle | 189 | 35 | 1.63 [0.57, 4.65] | 0.36 | 199 | 45 | 1.52 [0.60, 3.87] | 0.38 | ||
| CC: High | 401 | 50 | 1.34 [0.48, 3.79] | 0.78 | 412 | 61 | 1.25 [0.49, 3.16] | 0.64 | ||
|
| TT: Low | 24 | 4 | ref | — | 25 | 5 | ref | — | |
| AT: Middle | 184 | 34 | 1.25 [0.44, 3.56] | 0.68 | 193 | 43 | 1.10 [0.43, 2.81] | 0.84 | ||
| AA: High | 398 | 50 | 1.05 [0.37, 2.95] | 0.92 | 409 | 61 | 0.93 [0.37, 2.33] | 0.87 | ||
- —Alzheimer's Disease Neuroimaging Initiative (ADNI)
- —National Institute on Aging (NIA)10.13039/100000049
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLipoproteins and Cardiovascular Health · Alzheimer's disease research and treatments · HIV-related health complications and treatments
Introduction
1
Alzheimer's disease (AD) is the most common neurodegenerative disorder in late life, and its etiology is considered to involve both genetic susceptibility and environmental exposures. Among genetic factors, the apolipoprotein E (APOE) gene has been most consistently implicated. Multiple cohort studies have demonstrated that the APOE ε4 allele is a major genetic risk factor for AD, while the three common isoforms (APOE2, APOE3, and APOE4) influence both amyloid‐β–dependent and independent pathways implicated in AD pathogenesis [1, 2]. Although APOE is primarily a lipid regulating protein, our recent studies have shown that several proinflammatory proteins including C‐reactive protein (CRP) and MCP‐1 interact with APOE ε4 versus APOE ε2 in an opposite direction to impact AD risk [3, 4]. These findings suggest that APOE genotypes regulate both lipid metabolism and chronic inflammation differently to mediate AD risk. Together, these observations suggest a broader role of APOE in lipid and immune pathways, prompting the hypothesis that APOE may also be linked to proprotein convertase subtilisin/kexin type 9 (PCSK9), a lipid‐protein involved in lipid metabolism via the low density lipoprotein receptor (LDLR), the very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2) [5]. This hypothesis is further underscored by evidence that PCSK9 is also related to chronic inflammation, leading to the migration of immune cells through endothelia [6]. Recent studies have shown that cerebrospinal fluid PCSK9 levels are elevated in AD patients and correlate with APOE4 presence, indicating that ApoE4 carriers may be particularly susceptible to PCSK9‐mediated lipid dysregulation [7, 8]. This interaction could exacerbate amyloid‐beta accumulation, neuronal lipid imbalance, and neuroinflammation, highlighting the potential for APOE genotype to modulate responses to PCSK9‐targeted interventions. Basic research has discovered that PCSK9 is a key regulator of the AD pathological component Aβ clearance across the blood‐brain barrier (BBB) in the animal model [9]. In addition, PCSK9(‐/‐) mice showed increased levels of BACE1, the key protease in APP processing, and Aβ production and accumulation in the brain [10]. However, whether APOE genotypes interacting with PCSK9 for AD risk are unclear and understudied.
Since APOE protein regulates brain endothelia of BBB during peripheral inflammation [11], we hypothesized that blood PCSK9 impacts AD risk differently depending on the APOE genotypes. To address this hypothesis, we evaluated data from the Framingham Heart Study (FHS) cohort to examine the interactive effects of APOE genotypes and PCSK9 protein or polymorphisms for AD risk. We further used the data of the Alzheimer's Disease Neuroimaging Initiative (ADNI) study [12] to validated the findings.
Methods
2
This study was designed and reported in accordance with the CONSORT 2010 guidelines to ensure transparency, reproducibility, and completeness of reporting [13].
Participants
2.1
The first generation FHS cohort (Gen 1) was established in 1948, and the FHS Offspring Cohort started in 1971 was the second generation (Gen 2) of the original FHS cohort. Detailed information about FHS were previously reported [14]. Briefly, the FHS Offspring Cohort was strategically designed to extend and broaden the scope of the original study, delving deeper into the interplay of genetic, environmental, and lifestyle factors that influence cardiovascular health. The fifth wave of health exam (Core Exam 5) included 3799 participants (average age 55 years old, ranged 26–84) from 1991 to 1995, among which 1704 participants (age mean ± SD, 54.7 ± 9.88 years) had the data on PCSK9 protein and APOE genotype; in addition, 3048 participants (age mean ± SD, 54.5 ± 9.80 years) who had the genetic data on PCSK9 (SNPs genotype data from the TOPMed Project) and APOE genes. The exclusion citeria of the study were those missing APOE genotype, carrying APOE genotype ε2/ε4, baseline dementia when the protein was measured, and those who had follow‐up time < 3 years (Supporting Figure S1).
To validate our findings, cross‐validation analyses were performed using 1,476 participants from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (https://adni.loni.usc.edu). ADNI is a longitudinal, multicenter study designed to develop clinical, imaging, genetic, and biochemical biomarkers for the early detection and progression of Alzheimer's disease. For this analysis, logistic regression models were used to assess the association between PCSK9 genotypes and prevalent AD or all‐cause dementia, stratified by APOE ε4 carrier status. Results were reported as odds ratios (ORs) with 95% confidence intervals.
Ethical approval for this study was obtained from the Institutional Review Boards (IRBs) of Boston University Medical Center for the Framingham Heart Study (FHS) and from the respective IRBs overseeing the Alzheimer's Disease Neuroimaging Initiative (ADNI). All participants in the original cohorts provided written informed consent for study participation and data sharing. For the present secondary analysis, only deidentified and anonymized data were used, and no personally identifiable information was included. In accordance with FHS and ADNI policies and applicable IRB regulations, the use of deidentified data for secondary research purposes did not require additional informed consent.
PCSK9 Protein
2.2
The blood level of PCSK9 protein were measured by using the Aptamer Proteomic Profiling Lab Assay, and the detailed protocol was previusely reported [15]. In brief, the blood samples were collected in K2EDTA‐treated tubes in PMI patients and citrate‐treated tubes in FHS participants. Samples were centrifuged within 15 min at 2000g for 10 min to pellet cellular elements. The supernatant plasma was then aliquoted and frozen at −80°C. The SOMAscan™ proteomic profiling platform uses single‐stranded DNA aptamers that target 1,129 proteins. All assays were performed using SOMAscan reagents according to the manufacturer's detailed protocol [15].
PCSK9 SNPs
2.3
The NHLBI Trans‐Omics for Precision Medicine (TOPMed) program provided whole genome sequence (WGS), performed at several sequencing centers using DNA from blood. FHS WGS was conducted as part of TOPMed. Sequencing, performed at the Broad Institute of MIT and Harvard, achieved > ×30 depth of coverage. Details regarding the laboratory methods, data processing and quality control were previusely reported [16, 17]. Initially, all single nucleotide polymorphisms (SNPs) in the PCSK9 gene were identified. Genetic variants with minor allele frequencies of > 5% were used, and 3 top SNPs (rs502576, rs529787, and rs676297) were selected based on the association with PCSK9 protein (β > 0.20 and p < 0.001) in this study.
Demenita and AD
2.4
Dementia surveillance and diagnosis methods for FHS were previously published [3, 18]. In brief, participants with lower Mini‐Mental State Examinations (MMSE) scores were further evaluated by a comprehensive neuropsychological battery and subsequent neurological assessments. Participants flagged for possible cognitive impairment prompted by self‐ or family‐reported decline, physician referral, or review of medical records would also receive annual neurological and neuropsychological examinations.
A dementia review panel, consisting of a neurologist and a neuropsychologist, adjudicated all suspected cases. Dementia diagnoses were made according to DSM‐IV criteria, based on longitudinal cognitive assessments, caregiver interviews, medical records, neuroimaging, and autopsy data when available. Importantly, diagnoses of Alzheimer's disease dementia (ADD) were determined using NINCDS‐ADRDA criteria, thereby distinguishing ADD from preclinical AD or mild cognitive impairment (MCI).
Covariates
2.5
Covariates in our analyses include sex, age, education, body mass index (BMI; kg/m^2^), and medical histories gathered at the baseline. Educational attainment was categorized as no high school graduation, high school graduated, any college degrees, or > college degree. Cardiovascular diseases (CVDs) defined as incident coronary heart disease, stroke, heart failure, and peripheral arterial disease. The lipid profile, encompassing total cholesterol (TC), triglycerides (TG), HDL‐Cholesterol (HDL‐C), and calculated LDL‐Cholesterol (LDL‐C) using the Friedewald formula (LDL‐C = TC ‐ HDL‐C ‐ TG/5), was obtained from blood tests. Additionally, we computed the cumulative percentage of time on anti‐lipid medication use to gauge cumulative exposure to lipid‐lowering treatment ([number of core exams with medication use/nubmer of attended observation core examinations] × 100).
Statistical Analysis
2.6
Descriptive statistics were used to summarize baseline characteristics of the study population. Continuous variables were presented as means ± standard deviations or medians with interquartile ranges if the distribution was skewed. Categorical variables were summarized as frequencies and percentages (n, %). In case of missing data less than 10%, we assumed the data were missing at random and still employed complete case analysis in the multivariate analysis.
Dementia cases were defined as incident, new‐onset all‐cause dementia occurring after the baseline exam, with approximately 73% classified as AD dementia. Analyses included all participants at risk using the COX proportional hazards models to assess the association between the exposure variable (blood PCSK9 protein concentration or PCSK9 genotypes) and the follow‐up outcomes of the incident AD and all‐cause dementia. Adjusted analyses were conducted to control for potential confounders including age, sex, education, lipid profile and lipid treatments. The results were reported as hazard ratios (HR) with corresponding 95% confidence intervals (95% CI). Sensitivity analyses were performed to evaluate the robustness of our findings. The sensitivity analysis comprised stratified (subgroup) analyses and interaction testing.
Cross‐validation was performed using logistic regression model (the results were reported as odds ratio (OR) with its 95% CI) of the association between PCSK9 genotypes and prevlent of AD or all‐cause dementia among 1476 ADNI participants to validate the association between AD/Dementia and the selected SNPs of PCSK, stratified by APOE genotype.
All statistical analyses were conducted using R: A language and environment for statistical computing (version 4.3.1). Effect sizes are presented with 95% confidence intervals to convey both magnitude and precision. p values are reported following standard conventions (p < 0.001, to the nearest thousandth for 0.001–0.01, to the nearest hundredth for ≥ 0.01, and p > 0.99 where applicable). A two‐sided significance level of 0.05 was applied for all tests. All statistical analysis and reporting adhered to recommended standards for clinical research, following the Statistical Analyses and Methods in the Published Literature (SAMPL) guidelines and standard references [19].
Results
3
Characterization of the Samples Used for the Data Analyses
3.1
We used two datasets from the FHS Gen 2 cohort, exam 5, 1) the protein dataset was the one with the measurements of plasma PCSK9 protein (n = 1704) and 2) the genetic dataset was the one with the genetic data on PCSK9 gene (n = 3048) (Supporting Figure S1). Two datasets were compatible in basic characteristics including age (mean ± SD: 54.7 ± 9.88 vs. 54.5 ± 9.80 years), sex, education level, and APOE ɛ4 carriers (Supporting Table S1). In addition, the rate of CVD, the average BMI, lipid profiles and lipid treatment were similar between two datasets.
Characterization of the Relationship Between the Interactive Effects of APOE Genotypes and Plasma PCSK9 Protein for AD Risk
3.2
We first used the data on plasma PCSK9 from the dataset, which has the plasma protein concentrations measured by proteomics. There was no difference in plasma PCSK9 concentrations between APOE ɛ4 noncarriers and APOE ɛ4 carriers and among different APOE genotypes (Supporting Figure S2). Using unadjusted COX regression model (Table 1, model 1), we found an association between plasma PCSK9 and AD (HR [95%CI]: 0.85 [0.73, 0.99], p = 0.04). After adjusing for age, sex, education and APOE ɛ4, the association between plasma PCSK9 protein and AD risk became insignificant (Table 1, model 2). However, the interaction of APOE ɛ4 genotype and plasma PCSK9 protein was significantly associated with AD risk in the same COX regression model (HR [95%CI]: 1.59 [1.12, 2.27], p = 0.01, Table 1, Model 3). This interactive effect remained and strengthened after adding lipid profiles and lipid treatment in the COX model (HR [95%CI]: 1.80 [1.24, 2.61], p = 0.002, Table 1, Model 4). The similar interactive effects between APOE ɛ4 genotype and plasma PCSK9 were found for all‐cause dementia, where elevated peripheral PCSK9 protein is consistently associated with a reduced hazard of developing all‐cause dementia in the overall sample (e.g., Model 4: HR = 0.81 [0.66, 0.98], p = 0.033), and this association is strengthened when controlling for cardiovascular risk factors. Critically, similar to the AD findings, the protective association of higher PCSK9 is not present in APOE4 carriers; rather, in these individuals, the interaction between APOE4 and PCSK9 signifies increased risk (Model 4 interaction: HR = 1.65 [1.20, 2.28], p = 0.002) (Table 1). The data suggested that plasma PCSK9 impact AD dementia differently based on APOE ɛ4 noncarriers versus carriers, with similar patterns observed for both AD dementia and all‐cause dementia.
Next, we stratified the participants into APOE ɛ4 noncarriers and carriers. Among the noncarriers, plasma PCSK9 concentration was negative associated with AD risk after adjusting for age, sex, education, lipid profiles and lipid treatment (HR [95%CI]: 0.74 [0.58, 0.94], p = 0.01) (Table 2). In contrast, in APOE ɛ4 carriers, plasma PCSK9 concentration tended to be positively associated with AD risk but did not reach statistical significance. Similar tendencies of the associations were found for all‐cause dementia in an APOE genotype dependent manner (Table 2). Using the Kaplan‐Meier analyses, there was a tendency of low concentration of plasma PCSK9 increasing AD risk in in APOE ɛ4 noncarriers (p = 0.13) (Supporting Figure S3).
Characterization of the Relationship Between the Interactive Effects of the APOE and PCSK9 Genotypes for AD Risk
3.3
Next, we used the larger sample from FHS Gen 2 which has genetic information (n = 3048) (Supporting Figure S1 and Supporting Table S1). Among the PCSK9 polymorphisms, we chosed the top three PCSK9 gene SNPs, rs502576, rs676297 and rs529787, based on their influences on the plasma level of PCSK9 protein (β level > 0.20 in the linear regression model) (Supporting Table S2. These SNPs are all located in the 3rd intron of the PCSK9 gene (Supporting Figure S4). The 3 genotypes were significantly associated with plasma PCSK9 protein in the whole sample, especially in APOE ɛ4 noncarriers, but their influences on plasma PCSK9 protein in the presence of APOE ɛ4 were not significant (Supporting Figure S5). We combined APOE ɛ3/4 and APOE ɛ4/4 for the category of APOE ɛ4 because of the low numbers in APOE ɛ4/4 (Supporting Table S3 ).
Using COX model after adjusting for age, sex, education, lipid profiles and lipid treatment, we consistently found a negative association between those PCSK9 genotypes determining a low concentration of PCSK9 protein (Supporting Figure S5) and AD risk in APOE ɛ4 noncarriers (Table 3), which showed a similar trend of plasma PCSK9 protein and AD risk. Again these relationships were not found in APOE ɛ4 carriers. Using Kaplan‐Meier analyses, those genotypes determining a low concentration of PCSK9 protein in plasma were associated with a high risk of AD onset in the absence of APOE ɛ4 (Figure 1), but there were no associations between these SNPs and AD risk among those APOE ɛ4 carriers.
Kaplan‐Meier plot illustrating the survival analysis for Alzheimer's disease (AD) across different PCSK9 genotypes in the FHS study. The genotypes of three PCSK9 SNPs were categorized as “Low”, “Middle”, and “High” based on their association with blood PCSK9 protein levels in the APOE ε4 non‐carriers groups as shown in Supporting Figure S5, respectively. Survival analysis was conducted in the total sample (All), APOE ε4 non‐carriers (APOE4−), and APOE ε4 carriers (APOE4 +). Kaplan‐Meier curves for AD survival are shown for rs502576 (A) – CC (Low), GC (Middle), and GG (High); rs529787 (B) – GG (Low), CG (Middle), and CC (High); and rs676297 (C) – TT (Low), AT (Middle), and AA (High). Genotypes associated with lower PCSK9 protein concentrations exhibited a higher risk of AD development compared to other genotypes.
Validation of the Relationship Between the Interactive Effects of the APOE and PCSK9 Genotypes for AD Risk in the ADNI Study
3.4
We used the ADNI dataset and logistic model to validate the findings in FHS. After adjusting for age, sex and education, three PCSK9 polymorphisms, rs502576, rs676297, and rs529787, in the ADNI study were found to replicate the results in FHS (Table 3), e.g the PCSK9 SNPs were associated with AD dementia in APOE ɛ4 noncarriers, but not in APOE ɛ4 carriers (Supporting Table S4).
Since the basic research study discovered that PCSK9 acts as a key regulator of Aβ clearance [9] and the ADNI dataset has the data on the AD biomarkers in CSF, we examined the associations between the interactive effects of APOE ɛ4 and PCSK9 genotypes for the AD biomarkers in CSF. After adjusting for age, sex and education, we found that all three PCSK9 polymorphisms rs502576, rs676297, and rs529787, for a low concentrations of plasma PCSK9 had a lower level of CSF Aβ42, a biomarker indicating AD diagnosis, in APOE ɛ4 noncarriers, but not in APOE ɛ4 carriers (Figure 2). However, we did not find the differences of CSF total Tau (Supporting Figure S6) and pTau (Supporting Figure S7) across 3 PCSK9 genotypes of all three SNPs regardless of APOE ɛ4.
The boxplots for the CSF Aβ42 levels across the genotypes of PCSK9 gene stratified by APOE ε4 status in the ADNI study. Three SNPs in the PCSK9 gene were analyzed, with genotypes labeled as “Low”, “Middle”, and “High” based on corresponding blood PCSK9 protein levels in the APOE ε4 non‐carriers groups as shown in Supporting Figure S5, respectively. The genotypes were defined as follows: rs502576 (A) – CC (Low), GC (Middle), and GG (High); rs529787 (B) – GG (Low), CG (Middle), and CC (High); and rs676297 (C) – TT (Low), AT (Middle), and AA (High). CSF Aβ42 levels were compared across these genotype groups in the total sample (All), in APOE ε4 non‐carriers (APOE4−), and in APOE ε4 carriers (APOE4 +). The reported p‐values were from the non‐parametric Kruskal‐Wallis tests.
Discussion
4
Our study demonstrated a genotype‐specific relationship between PCSK9 and AD risk. Among APOE ε4 noncarriers, higher plasma PCSK9 levels were associated with a 26% (HR [95% CI]: 0.74 [0.58, 0.94]) lower risk of AD, and this protective association was consistent across both protein and genetic analyses. In contrast, among APOE ε4 carriers, neither plasma PCSK9 levels nor PCSK9 genotypes were significantly associated with AD risk (Table 2), indicating that the protective effect of PCSK9 is limited to noncarriers. Clinically, these findings suggest that noncarriers with higher PCSK9 may have substantially reduced AD risk, whereas ε4 carriers do not appear to benefit. Moreover, the beneficial association of higher circulating PCSK9 may extend beyond AD to reduce the risk of all‐cause dementia. The genotype‐dependent nature of this protective effect—restricted to non‐APOE ε4 carriers—likely reflects fundamental interactions between cholesterol/lipid pathways and APOE ε4–mediated neurodegeneration. The parallel risk patterns observed for both AD and all‐cause dementia reinforce the potential generalizability of PCSK9's neuroprotective role and highlight the importance of considering APOE ε4 status when evaluating therapeutic strategies or biomarker utility, particularly given the broad clinical and societal impact of all‐cause dementia.
As some proinflammatory proteins in blood impact APOE genotypes differently for AD risk [4, 20], PCSK9, a lipid related protein, also showed different or opposite associations with AD risk depending on APOE genotypes, e.g blood PCSK9 could be a protector for AD in APOE ɛ4 noncarriers but did not influence AD risk in APOE ɛ4 carriers. Consistent with our findings, other studies show the potential risks that may arise from low PCSK9 level (variant or inhibitor) for central nervous system (CNS) diseases including AD [21]. In the absence of APOE ɛ4 in humans, the genotypes associated with low concentrations of blood PCSK9 had a low level of Aβ42 in CSF, a diagnostic biomarker of AD, but this relationship did not appear among APOE ɛ4 carriers (Figure 2). This is consistent with the preclinical study that PCSK9(‐/‐) knockout mice showed increased levels of BACE1, leading to increased Aβ production and accumulation in the brain [10]. However, some studies investigate the central PCSK9 effects in the brain by using the AD mouse models with Aβ deposits in the brain with conflict results. For example, it was found that increased the brain expression of PCSK9 induced atherosclerosis, brain hypoxia and Aβ deposits in the brain [22]. In addition, knock‐down the expression of PCSK9 in the brain decreases Aβ level and neuroinflammation, and improve behavioral tests in these mice [23]. As our study focused on the effects of peripheral, but not central, PCSK9 for AD, the mechanism of peripheral PCSK9 for the brain is unclear. It is reported that PCSK9 plays a role in neurogenesis, neural cell differentiation, central LDL receptor metabolism, neural cell apoptosis, neuroinflammation, AD, alcohol use disorder, and stroke [24] that may explain the beneficial effects of peripheral PCSK9 on AD risk in the absence of APOE ɛ4.
The interactive effects with APOE ɛ4 noncarrier genotypes and increased plasma PCSK9 for reduced AD risk were influenced by adding lipid profile and anti‐lipid medications into the models (Tables 1 and 2). As some lipid species are beneficial for brain functions [25], the beneficial effects of peripheral PCSK9 on AD pathogenesis were probably through regulating lipid metabolism. Thus, diet components and taking the drugs targeting peripheral lipid metabolism could influence the associations between PCSK9 and AD. As taking the statin medications become common among midlife and old age in recent decades, lipid profiles in different populations have been changed and improved. The FHS cohort in this study only had < 5% of participants taking anti‐lipid medications in the 1990s, while the ADNI study was in early and the UKBB study was in late decade of 2000. A study using the UK Biobank (UKBB) cohort did not find that PCSK9, either protein or genotypes, is associated with AD risk [26].
While three genotypes were associated with blood PCSK9 concentrations in the absence of APOE ɛ4, the PCSK9 genotypes did not link with PCSK9 protein levels in the presence of APOE ɛ4 (Supporting Figure S5). APOE4 protein may suppress the expression of PCSK9 to inhibit PCSK9 regulating the pathways of immune responses and lipid metabolism. There was a trend of positive association between PCSK9 and AD risk in the presence of APOE4 (Table 2) but did not reach statistical significance (Tables 2 and 3; Figure 1) probably due to the lack of power. As lowering cholesterol in APOE4 carriers may decrease the risk of dementia based on other published literature [27, 28, 29, 30, 31, 32], it is necessary and probably safe for clinicians to treat hyperlipidemic patients who are APOE ε4 carriers with anti‐lipid medications including PCSK9 inhibitors.
PCSK9 inhibitors are the effective drugs to reduce the cholesterol levels and atherosclerosis to decrease the risk of cardiovascular diseases [5], but both cohorts we used did not have the data on PCSK9 inhibitor use. Up to date, the conclusions on PCSK9 inhibitors and AD risk are not conclusive. For example, in some trials, PCSK9 inhibitors including evolocumab do not show the effects on cognitive decline and complain in a large clinical trial [33, 34]. PCSK9 inhibitors may be harmful for AD in the minor frequencies of some PCSK9 genotypes [21, 24]. Another study did not show the interactive effects with APOE ɛ4 genotype for cognitive decline and complain in a large clinical trial [33]. In basic research, peripheral injection of PCSK9 inhibitors into the AD mouse models are shown to decrease Aβ level and neuroinflammation in the brain, and improve behavioral tests [9, 35].
While multiple factors including other genes, diets and anti‐lipid medication use can affect lipid metabolism and AD risk [36], our study did not account for diet as a potential confounding factor. Another limitation was the absence of experimental validation; however, future basic research is needed to investigate the mechanisms underlying the interaction between PCSK9 and APOE genotype in influencing AD risk. A third limitation was the lack of longitudinal PCSK9 measurements, which prevents drawing causal inferences regarding AD risk. Additionally, the lack of ethnic diversity of the FHS cohort may restrict the generalizability of our findings to more diverse populations. Note that residual confounding by age remains a potential limitation, as our primary analyses did not use age‐matched sampling. Future studies employing age‐matched replication analyses would help to confirm the robustness of these findings. A limitation of our study is that corrections for multiple comparisons were not applied, which may increase the risk of type I error. Nonetheless, the analyses were hypothesis‐driven and focused on predefined outcomes, which mitigates (but does not eliminate) this concern. Despite these limitations, the observed interaction between APOE genotypes and PCSK9 in our study may help explain some of the inconsistencies reported across different cohorts and clinical trials involving PCSK9 inhibitors in the context of AD. Clinical practice may need to avoid prescribing the PCSK9 inhibitors, but other anti‐lipid medications, for cardiovascular patients who are APOE ɛ4 noncarriers and carry the genotypes for low blood PCSK9 to reduce AD risk.
Author Contributions
Qiushan Tao: conceptualization, data curation, formal analysis, methodology, writing – original draft, writing – review and editing. Ting Fang Alvin Ang: conceptualization, data curation, formal analysis, methodology, supervision. Jinghan Huang: methodology. Indira Swetha Itchapurapu: project administration, Resources. Jesse Mez: writing – review and editing. Michael Alosco: writing – review and editing. Rhoda Au: conceptualization, funding acquisition, supervision, writing – review and editing. Lindsay A. Farrer: conceptualization, funding acquisition, supervision, writing – review and editing. Xiaoling Zhang: methodology, writing – review and editing. Wei Qiao Qiu: conceptualization, investigation, methodology, project administration, supervision, writing – original draft, writing – review and editing.
Ethics Statement
Ethical approval for this study was obtained through the respective Institutional Review Boards (IRBs) overseeing the Alzheimer's Disease Neuroimaging Initiative (ADNI) and the Framingham Heart Study (FHS). All human subjects in the original studies provided informed consent for participation and data sharing. For this secondary analysis, only deidentified, anonymized data were used, and no personally identifiable information (PII) was included. According to the policies of ADNI and FHS and in adherence with applicable IRB regulations, the use of such deidentified data for secondary research purposes did not require additional informed consent from participants.
Conflicts of Interest
The authors declare no conflicts of interest.
Transparency Statement
The lead author Wei Qiao Qiu affirms that this manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned (and, if relevant, registered) have been explained.
Supporting information
Supplemental Figure S1: The flow‐chart of inclusion and exclusion of the study participates. Supplemental Figure S2: The violin‐boxplot shows the plasma PCSK9 concentrations between the two groups, APOE4(+) (APOE ɛ4 noncarriers) versus APOE4(+) (APOE ɛ4 carriers) (A); among different APOE genotypes (B). Supplemental Figure S3: Kaplan‐Meier Plot Illustrating the Survival Analysis for Alzheimer's Disease (AD) in Relation to PCSK9 Protein Levels. Supplemental Figure S4: The LocusZoom plot of the three selected SNPs and their association with PCSK9 protein levels. Supplemental Figure S5: The boxplots of the blood PCSK9 protein levels across three PCSK9 genotypes in the absence and the presence of APOE ε4 allele. Supplemental Figure S6: The boxplots of the CSF total Tau levels among different genotypes of the three PCSK9 SNPs in the ADNI study at the baseline exams, stratified by APOE ε4 carriers' status. Supplemental Figure S7: The boxplot shows the CSF pTau levels among different genotypes of the three PCSK9 SNPs in the ADNI study at the baseline exams, stratified by APOE ε4 carriers' status. Supplemental Table S1: Baseline characteristics of two FHS datasets: the protein dataset and the genetic dataset. Supplemental Table S2: The association between PCSK9 protein level and the dosage of PCSK9 SNPs. Supplemental Table S3: The numbers of study subjects in the stratification of PCSK9 genotypes in the absence and the presence of APOE ε4 genotype. Supplemental Table S4: Stratification and logistic regression analyses for the association between PCSK9 genotypes and the AD or all‐cause dementia in ADNI study in the absence and the presence of APOE ε4 genotype.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1L. A. Farrer , “Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease: A Meta‐Analysis,” Journal of the American Medical Association 278, no. 16 (1997): 1349–1356.9343467 · pubmed ↗
- 2Y. Yamazaki , N. Zhao , T. R. Caulfield , C. C. Liu , and G. Bu , “Apolipoprotein E and Alzheimer Disease: Pathobiology and Targeting Strategies,” Nature Reviews Neurology 15, no. 9 (2019): 501–518.31367008 10.1038/s 41582-019-0228-7PMC 7055192 · doi ↗ · pubmed ↗
- 3Q. Tao , T. F. Alvin Ang , S. C. Akhter‐Khan , et al., “Impact of C‐Reactive Protein on Cognition and Alzheimer Disease Biomarkers in Homozygous Apolipoprotein E Ɛ4 Carriers,” Neurology 97 (2021): 1243–1252.10.1212/WNL.0000000000012512 PMC 848048434266923 · doi ↗ · pubmed ↗
- 4J. Huang , T. D. Stein , Y. Wang , et al., “Blood Levels of MCP‐1 Modulate the Genetic Risks of Alzheimer's Disease Mediated by HLA‐DRB 1 and APOE for Alzheimer's Disease,” Alzheimer's & Dementia 19, no. 5 (2023): 1925–1937.10.1002/alz.12851 PMC 1018218736396603 · doi ↗ · pubmed ↗
- 5S. Poirier , G. Mayer , S. Benjannet , et al., “The Proprotein Convertase PCSK 9 Induces the Degradation of Low Density Lipoprotein Receptor (LDLR) and Its Closest Family Members VLDLR and Apo ER 2,” Journal of Biological Chemistry 283, no. 4 (2008): 2363–2372.18039658 10.1074/jbc.M 708098200 · doi ↗ · pubmed ↗
- 6E. Punch , J. Klein , P. Diaba‐Nuhoho , H. Morawietz , and M. Garelnabi , “Effects of PCSK 9 Targeting: Alleviating Oxidation, Inflammation, and Atherosclerosis,” Journal of the American Heart Association 11, no. 3 (2022): e 023328.35048716 10.1161/JAHA.121.023328 PMC 9238481 · doi ↗ · pubmed ↗
- 7B. Papotti , M. Palumbo , M. P. Adorni , et al., “Influence of APOE 4 Genotype on PCSK 9‐lipids Association in Cerebrospinal Fluid and Serum of Patients in the Alzheimer's Disease Continuum,” Journal of Alzheimer's Disease 102, no. 1 (2024): 162–172.10.1177/1387287724128421339497318 · doi ↗ · pubmed ↗
- 8G. Testa , S. Giannelli , E. Staurenghi , et al., “The Emerging Role of PCSK 9 in the Pathogenesis of Alzheimer's Disease: A Possible Target for the Disease Treatment,” International Journal of Molecular Sciences 25, no. 24 (2024): 13637.39769398 10.3390/ijms 252413637 PMC 11727734 · doi ↗ · pubmed ↗