A novel ecotype of Anaplasma phagocytophilum complex in questing Ixodes fuscipes ticks
María L. Félix, Adriana Santodomingo, Richard Thomas, Diego Queirolo, Sebastián Muñoz-Leal, José M. Venzal

TL;DR
A new type of Anaplasma phagocytophilum bacteria was found in ticks in Uruguay, suggesting local transmission and expanding understanding of the species in South America.
Contribution
The discovery and genetic characterization of a novel A. phagocytophilum ecotype (VI) in Uruguay, the second in South America.
Findings
Anaplasma spp. DNA was detected in five Ixodes fuscipes nymphs from Uruguay.
Phylogenetic analysis confirmed a novel ecotype (VI) within the A. phagocytophilum clade.
The new ecotype is closely related to ecotype III and V, suggesting local host involvement in its transmission.
Abstract
Anaplasma phagocytophilum is a complex of tick-borne bacteria of medical and veterinary relevance, whose eco-epidemiology is well characterized in the Northern Hemisphere but remains poorly understood in South America. Here, we report in Uruguay the detection and genetic characterization of a novel A. phagocytophilum ecotype in South America. Questing Ixodes fuscipes, the only member of the Ixodes ricinus complex in the country, were collected in five localities in Uruguay, and the presence of Anaplasma spp. DNA was assessed using PCR to amplify fragments of the 16S ribosomal RNA (rrs), gltA and groEL genes. A total of 223 Ixodes fuscipes ticks were collected between 2017 and 2022 in five localities. PCR screening and subsequent sequencing identified Anaplasma spp. DNA in five nymphs from the Rivera and Tacuarembó departments. Phylogenetic analyses of rrs, gltA and groEL sequences of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1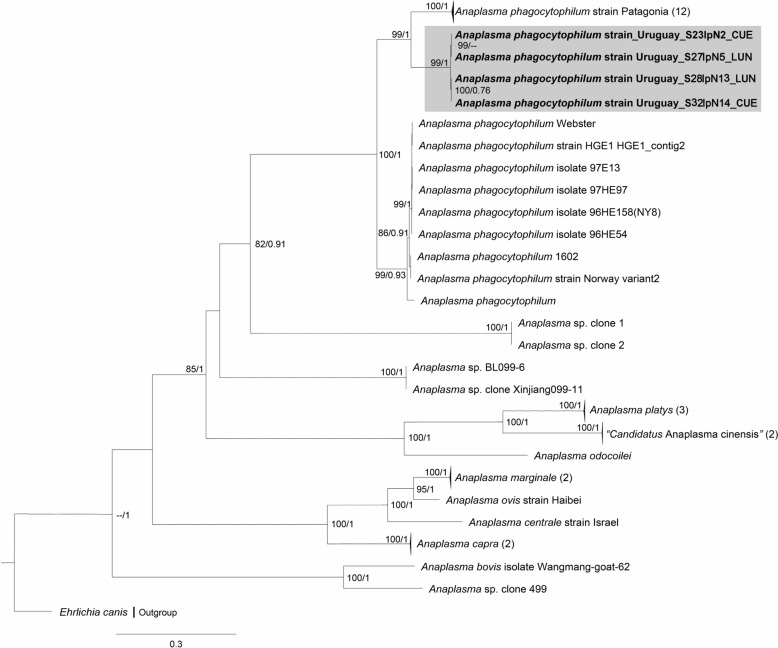 Figure 2
Figure 2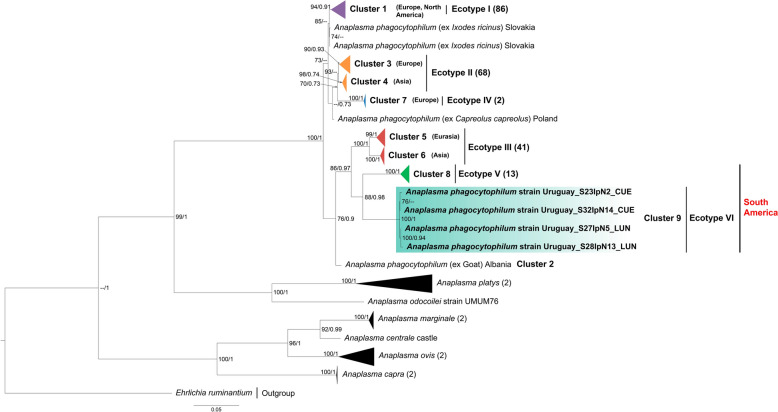 Figure 3
Figure 3 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsVector-borne infectious diseases · Bartonella species infections research · Invertebrate Immune Response Mechanisms
Background
The genus Anaplasma (Rickettsiales: Anaplasmataceae) comprises small, pleomorphic, Gram-negative bacteria that thrive in a variety of vertebrate hosts and which are primarily transmitted by ixodid ticks (Acari: Ixodidae) [1, 2]. The systematics of genus Anaplasma currently comprises six formally recognized species (A. bovis, A. centrale, A. marginale, A. ovis, A. phagocytophilum and A. platys), along with two provisional species (A. odocoilei and A. capra) and several additional candidate species and genovariants [3, 4].
The most widespread and genetically diverse species within genus Anaplasma is A. phagocytophilum, which poses a significant threat to human and animal health [3, 5]. This species is the etiological agent of human granulocytic anaplasmosis (HGA), tick-borne fever in domestic ruminants and granulocytic anaplasmosis in horses, dogs and cats [6]. Its natural enzootic cycles are well-described in the Northern Hemisphere, and its vertebrate hosts involve wild ruminants, rodents, insectivores, carnivores, raccoons, wild boards and, less frequently, birds and reptiles [3]. The primary vectors include the ticks Ixodes ricinus in Europe, Ixodes persulcatus in Eurasia and Ixodes scapularis and Ixodes pacificus in North America, where they have been recorded transmitting infections to humans and animals [5, 7, 8]. The ability of A. phagocytophilum to infect a broad spectrum of hosts points to its ecological plasticity and epidemiological significance [3].
Multiple strains and genovariants of A. phagocytophilum have been described, often linked to specific epidemiological cycles, host preferences and variable clinical pictures [9–11]. Molecular markers, such as the 16S ribosomal RNA (rrs), ankyrin A (ankA), major surface protein 4 (msp4), citrate synthase (gltA) and the heat-shock operon (groEL) genes, have been widely employed to disentangle this diversity [3, 4, 9, 12]. Among these, groEL stands out not only as the most informative marker for distinguishing A. phagocytophilum lineages but also as the locus with the most comprehensive sequence dataset, classified into four ecotypes that subdivide into seven phylogenetic clusters across Europe, Asia and North America [3, 4].
A bacterial ecotype is defined as a monophyletic group of strains sharing a similar ecological niche, for which between-ecotype sequence divergence is significantly greater than within-ecotype divergence for a given gene [13, 14]. In the case of A. phagocytophilum, ecotypes and phylogenetic clusters have been delineated based on groEL genetic divergence, geographic distribution, enzootic cycles, host range and pathogenic potential [3, 4, 9, 15]. For example, ecotypes I and II are strongly associated with Ixodes spp. ticks parasitizing cervids in Europe, ecotype III is linked to small mammals (rodents and insectivores) and their Ixodes spp. vectors in Eastern Europe and parts of Asia, whereas ecotype IV is associated with birds and their avian ticks, mainly in Europe [3]. Importantly, A. phagocytophilum strains belonging to ecotype I are zoonotic and include variants that cause HGA [3].
As key hosts in tick–pathogen systems, cervids sustain adult Ixodes spp. ticks, some of which can transmit Anaplasma spp., and they are also recognized as competent reservoir hosts for these infections [16]. In parallel, small mammals and birds contribute by feeding on larval and nymphal stages, thereby ensuring the continuity of Ixodes spp. life-cycles and the persistence of enzootic cycles of pathogens such as Anaplasma spp. Several Anaplasma spp. have been documented in South America [17], including the genetic detection and evidence of exposure to A. phagocytophilum in a variety of wild hosts [18–22]. However, ecotypes of this species were not described until in 2023, when Santodomingo et al. [4] characterized a novel ecotype of A. phagocytophilum (ecotype V) in wild pudu cervids (Pudu puda) and Ixodes stilesi ticks in Chile.
More recently, a study identified the occurrence of ecotype I in dogs in the Galapagos Islands, Ecuador [23]. Despite the current geographic restrictions associated with A. phagocytophilum ecotypes, these findings shed light on the epidemiology and systematics of the species beyond the Northern Hemisphere, revealing that South America may harbor a broader diversity of A. phagocytophilum ecotypes than previously recognized, with endemic lineages sustained by local host–vector assemblages. Indeed, South America presents ecological conditions capable of sustaining enzootic cycles of Anaplasma species, with its high diversity of Ixodes spp. ticks and wildlife hosts providing a natural setting favorable for their circulation and persistence [17, 24].
Uruguay has the environmental conditions, host–vector assemblages and an epidemiological context that make it a suitable setting for exploring Anaplasma species diversity [24, 25]. Records of three species (A. platys, A. centrale, and A. marginale) [26–28] and one lineage (Anaplasma sp. genotype Mazama) [25] highlight both the diversity already present and the need to further investigate hidden enzootic cycles involving other species, such as A. phagocytophilum. Accordingly, this study aimed to detect and characterize Anaplasma spp. using markers for ribosome- and protein-encoding genes, in questing Ixodes fuscipes ticks, the sole representative of the Ixodes ricinus complex in the country.
Methods
Study area
Sampling campaigns were conducted in five Uruguayan localities between September 2017 and May 2022. Four of these localities are situated in the northern region of the country: Arroyo Sepulturas, in Artigas Department (− 30.851389, − 56.072500); Lunarejo, in Rivera Department (− 31.141389, − 55.900278); Gruta de los Cuervos, in Tacuarembó Department (− 31.618889, − 56.046389); and Puntas de Arapey, in Salto Department (− 31.156944, − 56.140556). The fifth site, Laguna Negra, in Rocha Department (− 34.085833, − 53.738056), is found in the southeast of the country.
Collection of samples
Ticks were collected on predetermined transects using the flagging method, which consists of dragging a white cloth measuring 1.20 × 0.80 m across the vegetation. Depending on the site, sampling covered an area of at least 750 to 1000 m^2^ and was conducted by two collectors. Ticks were picked up from the cloth every 5–10 m and preserved in tubes containing 95% ethanol (Sigma-Aldrich®, St. Louis, MO, USA) until laboratory processing [29, 30].
Laboratory proceedings
Tick identification
Ticks were identified following the morphological descriptions and keys of Venzal et al. [31] and Nava et al. [24], using a Nikon SMZ1000 stereo microscope (Nikon Corp., Tokyo, Japan). To support morphological identification, we molecularly characterized one tick from each sampling site by sequencing fragments of the cytochrome* c* oxidase subunit I (cox1), amplified following Folmer et al. [32]. Primer sequences and PCR conditions are given in Table 1. Table 1. Primers and thermal conditions used for PCR detection and genetic characterization of Anaplasma phagocytophilum and Ixodes fuscipesOrganismGene^a^PCRPrimerSequenceTo ©°)Expected length (bp)ReferencesTickscox1PrimaryLCO1490GGTCAACAAATCATAAAGATATTGG50710[32]HCO2198TAAACTTCAGGGTGACCAAAAAATCAAnaplasmataceae16S rRNAPrimaryEHR16SDGGTACCYACAGAAGAAGTCC55345[34]EHR16SRTAGCACTCATCGTTTACA GCPrimaryfD1AGAGTTTGATCCTGGCTCAG55 ~ 1500[66]rP2ACGGCTACCTTGTTACGACTTAnaplasma spp.groElPrimaryHS1aAITGGGCTGGTAITGAAAT48 ~ 1400[67, 68]HS6aCCICCIGGIACIAIACCTTCNestedHS43ATWGCWAARGAAGCATAGTC551297[69]HSVRCTCAACAGCAGCTCTAGTAGC551297[69]PrimaryEEgro1FGAGTTCGACGGTAAGAAGTTCA55670[70]Anagro712RCCGCGATCAAACTGCATACCPrimaryAnagro122FAAATACGGTWGTCACGGG55385[71, 72]Anagro649RCTTTCTTCRACAGTTATAAGPrimaryAnaGroe240FATTAGYAAGCCTTATGGGTC55432AnaGro712RCCGCGATCAAACTGCATACCPrimaryAnaplatF2GCGTAGTCCGATTCTCC AGT59650[71, 73]AnaGro712RCCGCGATCAAACTGCATACCPrimarygro607FaGAAGATGCWGTWGGWTGTACKGC57664[74]gro1294RbAGMGCTTCWCCTTCWACRTCYTCNestedgro677FbATTACTCAGAGTGCTTCTCARTG55315[74]gro1121RbTGCATACCRTCAGTYTTTTCAACPrimaryAnagroUruFATTAGYAAGCCTTATGGRTC551072This studygroE-1236asTCTTTRCGTTCYTTMACYTCAACTTC[75]Anaplasma spp.gltAPrimaryEHR-CS131FCAGGATTTATGTCTACTGCTGCTTG541048[76]EHR-CS1226RCCAGTATATAAYTGACGWGGACGPrimaryF4bCCAGGCTTTATGTCAACTGC55800[77]R1bCGATGACCAAAACCCATNestedEHR-CS136FTTYATGTCYACTGCTGCKTG55650 [77]EHR-CS778RGCNCCMCCATGMGCTGG^a^cox1 Cytochrome* c* oxidase subunit I, gltA citrate synthase, groEL heat-shock operon, rrs 16S ribosomal RNA
DNA isolation and Anaplasmataceae detection
Since the transmission of A. phagocytophilum relies mainly on horizontal transfer between ticks and vertebrate hosts, as well as on transstadial transmission in its vector [12], this study focused on nymphs and adult ticks.
Prior to DNA extraction, all collected ticks (kept in tubes containing 95% ethanol) were rinsed with distilled water for 10 min to remove ethanol and then sectioned longitudinally. Genomic DNA was isolated using the GeneJET Genomic DNA Purification Kit (Thermo Fisher Scientific Baltics, Vilnius, Lithuania), according to manufacturer’s instructions. DNA was eluted in 100 μl of buffer (10 mM Tris–HCl, pH 9.0, 0.1 mM EDTA), following which DNA concentration was quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and the quality of the DNA sample was checked by assessing the A260/A280 absorbance ratio. Samples with absorbance ratios between 1.6 and 2.0 were accepted as suitable for PCR amplification protocols [33]. Extracted DNA from ticks was also tested by amplifying the cox1 gene as an internal control.
Anaplasma spp. detection was achieved using primers EHR16SD/EHR16SR to amplify a short fragment of the rrs gene (short-rrs) [34]. For full genetic characterization, samples that tested positive in the screening assay underwent distinct PCR protocols targeting long fragments of the rrs (long-rrs), gltA and groEL genes, as detailed in Table 1. DNA of A. marginale isolate Uru 61 (GenBank accession number OP383022) was used as the positive control, and ultrapure water served as the no-template control. Each PCR was performed in 25 μl of reaction mixture (2 μl of each primer [0.4 μM], 4.5 μl of ultrapure water, 12.5 μl of MangoMix [Bioline, Memphis, TN, USA] and 4 μl of template DNA., in a SimpliAmp™ thermal cycler (Applied Biosystems, Thermo Fisher Scientific)
PCR amplicons were verified by electrophoresis in a 1.5% agarose gel, loading 5 μl of product stained with GoodView Nucleic Acid Stain (Beijing SBS Genetech Co. Ltd., Beijing, China) and visualized with a CSLUVTSDUO312 ultraviolet (UV) transilluminator (Cleaver Scientific Ltd., Rugby, UK). Amplicons of the expected size were purified using the GeneJET PCR Purification Kit (Thermo Fisher Scientific Baltics) and subsequently submitted to Macrogen (Seoul, South Korea) for bidirectional Sanger sequencing.
Genetic analysis
Assembly and sequence analyses
Consensus sequences were generated from AB1 files in Geneious Prime v2021.2.2 (www.geneious.com) using a Phred score threshold ≥ 20 during base calling [35, 36]. To identify orthologous sequences, we used the basic local alignment search tool (BLASTn) (https://blast.ncbi.nlm.nih.gov) to compare consensus sequences against the non-redundant GenBank nucleotide database (https://www.ncbi.nlm.nih.gov).
Phylogenetic analysis
Phylogenies were performed under maximum-likelihood (ML) [37] and Bayesian inference (BI) [38, 39] approaches, following the phylogenetic frameworks outlined by Santodomingo et al. [4] to resolve the evolutionary relationships of Anaplasma spp. based on long-rrs, gltA and groEL sequences, together with orthologous sequences identified through BLASTn searches. Alignments for each marker were constructed with the Multiple sequence alignment program MAFFT v7 algorithm using default parameters [40] and subsequently curated using the Block Mapping and Gathering with Entropy (BMGE) v1.1 under predefined parameters [41].
ML and BI trees were inferred in IQ-TREE v1.6.12 [42] and MrBayes v3.2.6 [43], respectively. Protein-encoding gene datasets (gltA and groEL) were partitioned into codon positions as described by Santodomingo et al. [4]. ML best-fitting substitution models and best-partition scheme for protein-encoding datasets were determined with ModelFinder command “TESTNEWONLYMERGE -mrate G” [44]. A non-protein-coding 16S rRNA gene best-fit evolutionary model was computed using the ModelFinder command “-m TESTNEWONLY -mrate G.” The reliability of the ML tree topologies was evaluated through hill-climbing strategies combined with a stochastic perturbation procedure and further supported by 1000 ultrafast bootstrap (UFBoot) replicates [45].
BI best evolutionary models and phylogenies were inferred using the MrBayes commands “lset nst = mixed rates = gamma” and “lset = mixed rates = invgamma” for non-coding and protein-encoding datasets, respectively [43, 46]. The robustness of the inferred BI tree was evaluated by sampling trees every 1000 generations, with the first 25% as burn-in, implementing four Markov chain Monte Carlo (MCMC) chains through two independent tests of 40 × 10^6^ generations. The correlation and effective sample size (ESS) of the MCMCs were confirmed using Tracer v1.7.1 [47]. All best-fit models were selected according to the Bayesian information criterion (BIC) [48]. Trees were visualized and edited with FigTree v1.4.1 (http://tree.bio.ed.ac.uk/software/figtree/) and Inkscape v1.3.1 (https://inkscape.org/es/). The final tree for each marker integrates the consensus topologies from ML and BI analyses, constructed as described by Santodomingo et al. [49].
Genetic distance analysis
Average sequence divergence within and among A. phagocytophilum ecotypes was assessed using the groEL sequences generated in the present study, in combination with the groEL dataset proposed by Santodomingo et al. [4], which comprises 214 sequences with > 70% coverage and uses Anaplasma odocoilei and A. platys as outgroups (218 sequences in total). The alignment was generated using the multiple sequence alignment program MAFFT with default settings. Subsequently, the corrected pairwise distance was assessed with raxmlGUI [50, 51] in RAxML v8 [52] with the GTR + GAMMA + I substitution model.
Results
Tick collection and identification
In total, 223 ticks were collected across five sampling localities, consisting of 19 adults (10 females and 9 males) and 204 nymphs (see Table 2 for details of tick life stage per locality). Morphological identification confirmed that all specimens correspond to I. fuscipes. DNA extractions yielded high-quality DNA, and PCR amplification of the tick cox1 produced amplicons of the expected size (approx. 710 bp) in all samples, validating the success of the DNA extractions. cox1 sequences obtained from one tick specimen per sampling site after BLASTn analyses showed 98.98–100% identity to I. fuscipes isolate IF URUI previously characterized in Uruguay, thus supporting the morphological identification of the specimens (Additional File 1: Table S1). Comparisons also showed 90.61–90.82% identity with Ixodes pararicnus and 91.65–92.26% with Ixodes chacoensis sequences, both members of the I. ricinus complex in the Southern Cone of South America. Table 2. Metadata collected from Ixodes fuscipes ticks and samples that tested positive for Anaplasma phagocytophilumCollection site (total no. of ticks)Collection dateDevelopmental stageNumber of ticksPositive samples (n) / codeGenBank accession numberrrsgltAgroELGruta de los Cuervos, Tacuarembó (88)September 2017Female10Nymph70December 2017Nymph100January 2018Nymph31 / S23IpN2(42)PX418211PX394610PX394611March 2018Nymph60May 2018Nymph30June 2018Male10Nymph90July 2018Female10Nymph30July 2021Male20Female30Nymph101 / S32IpN14(124)PX418208PX394607PX394612September 2021Nymph10November 2021Male10Female10Nymph10February 2022Nymph50May 2022Male10Nymph190Lunarejo, Rivera (106)September 2017Male20Nymph70November 2017Nymph80December 2017Nymph50March 2018Nymph10May 2018Nymph70June 2018Male10Nymph61 / S27IpN5(65)PX418209PX394609PX394613July 2018Nymph131 / S28IpN13(90)PX418212PX394608PX394614September 2021Nymph120November 2021Nymph60December 2021Nymph171 / S36IpN14(174)PX418210--February 2022Nymph30May 2022Nymph180Arroyo Sepulturas, Artigas (11)July 2021Female10Nymph40October 2021Female10Nymph40November 2021Nymph10Puntas de Arapey, Salto (2)July 2021Female10Male10Laguna Negra, Rocha (16)November 2020Female10Nymph150Total2235 gltA Citrate synthase, groEL heat-shock operon, rrs 16S ribosomal RNA
Anaplasma spp. detection and genetic analysis
Of the 223 ticks screened for Anaplasma spp., five ticks tested positive in the short-rrs PCRs, including two nymphs from Tacuarembó (2.27%) and three from Rivera (2.87%), producing amplicons of approximately 345 bp. Subsequent PCRs assays for full genetic characterization generated five sequences for long-rrs (742–1432 bp) and four sequences each for the gltA (579–628 bp) and groEL (983–1,035 bp) genes (Additional file 2: Table S2). BLASTn comparisons of the long-rrs genotypes revealed 98.16–99.80% identity with A. phagocytophilum sequences previously characterized in Cervus elaphus (OR268760) in the UK, a dog in South Africa (MK814402), a goat in China (HQ872464) and a human in South Korea (CP035303) (Additional file 2: Table S2). For gltA, pairwise analyses showed 85.52–86.08% identity with A. phagocytophilum isolate IS21 (OP585592) obtained from I. stilesi ticks in Chile (Additional file 2: Table S2). In contrast, groEL sequences displayed 93.43–93.59% identity with A. phagocytophilum isolate 21F-2 (MT018452) from Marmota himalayana in China (Additional file 2: Table S2).
Phylogenetic and pairwise genetic analyses
Phylogenies inferred from the three loci (long-rrs, gltA, and groEL) consistently placed Anaplasma spp. genotypes characterized from I. fuscipes ticks within the A. phagocytophilum clade, forming a monophyletic group with the A. phagocytophilum Patagonia strain (Figs. 1, 2, 3). For the groEL gene, the Uruguayan sequences formed a distinct, well-supported lineage (Cluster 9) together with A. phagocytophilum strain Patagonia, both closely related to ecotype III (Fig. 3). Cluster 9 was further supported by genetic distance analyses, as average sequence divergences within the Uruguayan cluster were consistently lower than those observed among ecotypes (Table 3).Fig. 1. Maximum likelihood (ML) and Bayesian inference (BI) consensus tree of the long 16S ribosomal RNA gene (rrs) for Anaplasma species, highlighting the phylogenetic placement of the Anaplasma phagocytophilum “Uruguay” strain, shown within the gray box. Phylogenetic analyses incorporated 49 Anaplasma spp. sequences (alignment length: 1384 bp). Best-fit evolutionary models calculated for ML and BI methods were TPM3u + F + G4; and M85, M134, M177, M179, M147, M200, respectively. Only nodes with ultrafast bootstrap values > 70% for ML [45] and Bayesian posterior probabilities ≥ 0.71 for BI [65] are displayed above or below branches. Numbers in brackets indicate the number of sequences in each collapsed clade. The scale bar represents nucleotide substitutions per site. Sequence details and GenBank accession numbers are provided in Additional file 3: Table S3Fig. 2Maximum likelihood (ML) and Bayesian inference (BI) consensus tree of the citrate synthase gene (gltA) for Anaplasma species, highlighting the phylogenetic placement of the Anaplasma phagocytophilum “Uruguay” strain, shown within the gray box. Phylogenetic analyses incorporated 43 Anaplasma spp. sequences (alignment length: 1026 bp). Best-fit evolutionary models calculated for ML and BI methods were GTR + F + G4 (position-1), TIM3 + F + G4 (position-2), TPM2u + F + G4 (position-3); and M64, M173, M175, M171, M125 (position-1); M80, M135, M164, M145, M166 (position-2); M90, M177, M152, M183, M136 (position-3), respectively. Only nodes with ultrafast bootstrap values > 70% for ML [45] and Bayesian posterior probabilities ≥ 0.71 for BI [65] are displayed above or below branches. Numbers in brackets indicate the number of sequences in each collapsed clade. The scale bar represents nucleotide substitutions per site. Sequence details and GenBank accession numbers are provided in Additional file 3: Table S3Fig. 3Maximum likelihood (ML) and Bayesian inference (BI) consensus tree of the heat-shock operon gene (groEL) for Anaplasma species, highlighting the phylogenetic placement of the Anaplasma phagocytophilum “Uruguay” strain, shown within the turquoise box. Phylogenetic analyses incorporated 230 Anaplasma spp. sequences (alignment length: 1224 bp). Best-fit evolutionary models calculated for ML and BI methods were TIM + F + G4 (position-1); TN + F + G4 (position-2); and K3Pu + F + G4 (position-3); and M45, M136, M142, M130, M185, M139 (position-1); M81, M40 (position-2); M15, M50, M85, M122 (position-3), respectively. Only nodes with ultrafast bootstrap values > 70% for ML [45] and Bayesian posterior probabilities ≥ 0.71 for BI [65] are displayed above or below branches. Ecotype colors (I–V) follow Jaarsma et al. [12] and Santodomingo et al. [4]. Numbers in brackets indicate the number of sequences in each collapsed clade. The scale bar represents nucleotide substitutions per site. Sequence details and GenBank accession numbers are provided in Additional file 4: Table S4Table 3Average sequence divergence within ecotypes (highlighted in italics) and among ecotypes calculated according to the corrected pairwise distance for the groEL gene (alignment length: 1035 bp) of Anaplasma phagocytophilumEcotypeEcotype IEcotype IIEcotype IIIEcotype IVEcotype VEcotype VIEcotype I0.004930Ecotype II0.0175870.003021Ecotype III0.0559140.0503610.002056Ecotype IV0.0401540.0301810.0564960.000001Ecotype V0.0860590.0776650.0790010.0782040.003102Ecotype VI0.0781100.0785940.0690030.0792600.0796960.002135
Discussion
Based on phylogenies of the long-rrs, gltA and groEL genes, we identified an uncharacterized genovariant of A. phagocytophilum associated with I. fuscipes ticks, which we propose to designate as “Uruguay” variant (Figs. 1, 2, 3). Divergence values of partial groEL sequences further support its recognition as a novel ecotype within the A. phagocytophilum complex (ecotype VI) (Table 3), representing the first ecotype reported in Uruguay and the second ecotype characterized in South America. Notably, the groEL phylogeny provided strong evidence for regional differentiation, with the Uruguayan genovariant forming a distinct and well-supported lineage (Cluster 9) together with the strain Patagonia (ecotype V, Cluster 8) (Fig. 3). The detection of A. phagocytophilum in I. fuscipes aligns with its membership in the I. ricinus complex, a group that includes species such as Ixodes scapularis and I. ricinus, both well recognized for their involvement in Anaplasma spp. enzootic cycles in the Northern Hemisphere [3, 53].
The detection of ecotype VI in Uruguay, alongside ecotype V previously reported in pudu deers and I. stilesi ticks in Chile [4], supports the existence of multiple native A. phagocytophilum lineages circulating in the Southern Cone of South America. In contrast, the recent report of ecotype I in dogs in the Galápagos Islands [23] likely reflects an introduction rather than an natural endemic cycle. Collectively, these findings suggest that A. phagocytophilum is not restricted to recent introductions from the Northern Hemisphere but also includes native lineages that circulate in local wildlife-tick systems, maintained through distinct enzootic cycles under local ecological conditions. Ecotypes V and VI clearly contrast with their northern counterparts, differing not only phylogenetically (Fig. 3) and geographically but also in terms of the identity of their possible vectors and vertebrate hosts. These patterns point to the role of local host–vector assemblages in shaping A. phagocytophilum diversity in South America [4].
In the Northern Hemisphere, the eco-epidemiology of the main A. phagocytophilum ecotypes (I and II) is well characterized: cervids act as major reservoirs for both ecotypes while supporting adult Ixodes spp. ticks, whereas small mammals can also contribute to their maintenance by hosting larval and nymphal stages, thereby ensuring the continuity of enzootic cycles [3, 8]. Birds also play a role in maintaining immature stages of Ixodes spp., and are especially involved in the circulation of ecotype IV, which is associated with ornithophilic vectors such as Ixodes frontalis and Ixodes ventalloi; however, their contribution to the persistence of ecotypes I and II appears to be more limited and secondary [3].
Variants of A. phagocytophilum show host and vector specificity, leading to the maintenance of independent enzootic cycles that reflect adaptation to distinct ecological niches [3, 9]. From an ecological perspective, the recognition of “Uruguay” genovariant as an additional A. phagocytophilum ecotype associated with I. fuscipes points to a local transmission cycle in Uruguay, likely involving the vertebrates on which these ticks feed as potential reservoir hosts [3, 4]. Due to the close phylogenetic relationship between ecotype VI and ecotype V (Fig. 3), it cannot be ruled out that both lineages share similar eco-epidemiological strategies, including the involvement of deer in their maintenance. In this context, the Gray brocket deer (Subulo gouazoubira), the only cervid recorded as the main host of adult I. fuscipes [54], could be involved in the cycle of this new ecotype, although this hypothesis requires evaluation in future studies. It should be noted, however, that no adult ticks tested positive in this study, and given the small sample size (n = 19), broader sampling will be required to assess the potential role of this life stage in the enzootic cycle of ecotype VI.
Although A. phagocytophilum is primarily transmitted by Ixodes spp. [3], the mere detection of its DNA in I. fuscipes is insufficient evidence to assume a vectorial role [55]. Since Ixodes species feed on up to three different hosts during their life-cycle [56], the detected DNA could reflect residual, undigested blood of a previously infected host rather than an infection in the tick itself. Importantly, all ticks that tested positive in this study were nymphs collected from vegetation, and none of these showed signs of engorgement. Ultimately, confirmation of vector competence will require additional evidence, such as successful pathogen isolation, detection of A. phagocytophilum in salivary glands or experimental transmission to vertebrate hosts [55].
Ixodes species themselves have not been recognized as reservoirs of A. phagocytophilum [3], and transovarial transmission has only been documented in Dermacentor albipictus ticks [57]. The detection of positive nymphs in our study suggests that immature I. fuscipes likely acquire the bacterium from vertebrate hosts occurring in the localities of Lunarejo (Rivera Department) and Gruta de los Cuervos (Tacuarembó Department). Small mammals and birds are well established as primary hosts for larval and nymphal stages of Ixodes spp. [56], and in South American countries, immature I. fuscipes have been recorded parasitizing rodents such as Akodon azarae and Oligoryzomys nigripes, as well as birds, including Phacellodomus striaticollis, Syndactyla rufosuperciliata, Turdus rufiventris and Turdus albicollis [54].
Notably, ecotypes V (Patagonia) and VI (Uruguay) cluster together and are closely related to ecotype III (Fig. 3), which involves small mammals and their Ixodes spp. ticks in its enzootic cycles. This phylogenetic proximity suggests that the South American ecotypes (V and VI) may likewise rely on small mammals for their maintenance. Taken together, observed host associations, their overlapping distributions in Tacuarembó and Rivera Departments [54, 58, 59] and the phylogenetic link between ecotypes V, VI and III highlight plausible vertebrate hosts candidates for involvement in the transmission cycles of A. phagocytophilum. Future investigations should test this hypothesis through targeted sampling of both vertebrate hosts and I. fuscipes ticks to detect A. phagocytophilum in these and other regions where the species occurs. Meanwhile, the epidemiological cycle of A. phagocytophilum genovariant “Uruguay” remains undetermined.
Beyond Uruguay, several rrs genotypes associated with A. phagocytophilum have been reported in South American mammals, including deer (S. gouazoubira), rodents (Cavia sp. and Calomys cerqueirai), peccaries (Tayassu pecari and Dicotyles tajacu), sloths (Bradypus tridactylus) and coatis (Nasua nasua) [19–22], as well as in South American birds such as black vultures (Coragyps atratus), Orinoco geese (Oressochen jubatus), dusky-legged guans (Penelope obscura), caracaras (Caracara plancus) and Magellanic penguins (Spheniscus magellanicus) [60–63]. While these reports provide valuable preliminary insights, most detections relied on short rrs fragments (382–544 bp), lacked corresponding gltA or groEL sequences and were based on limited taxon sampling for phylogenetic analyses. As BLASTn comparisons did not reveal matches between the rrs sequences reported in those studies and the present study, and given that phylogenetic inferences in Anaplasma spp. require sufficiently long rrs, gltA, and groEL sequence fragments [3, 4, 64], complemented by dense taxon sampling of both ingroup and outgroup taxa to delimit lineages and evaluate their monophyly and evolutionary relationships with higher confidence [4], we did not include those sequences in our analyses.
Finally, this study has a number of limitations worth highlighting. First, the low number of positive nymphs (n = 5) restricts any evaluation of both the epidemiological significance of the findings (including bacterial prevalence and enzootic cycle stability) and the assessment of genetic variability within the detected lineage. Second, pathogen detection relied solely on DNA recovered from ticks, with functional evidence of infection or transmission lacking, which prevents the role of I. fuscipes as a vector. Third, the absence of vertebrate host analysis from surveyed areas limited our capacity to identify potential host reservoirs and to approximate the enzootic cycle. Consequently, the ecological and epidemiological interpretations presented here should be considered to be preliminary.
Conclusions
The eco-epidemiology of A. phagocytophilum in South America remains less well understood than that in the Northern Hemisphere. Based on groEL divergence criteria typically applied for ecotype delimitation, this lineage shows genetic differentiation comparable to those observed among established ecotypes. In this context, our study documents a novel ecotype (ecotype VI, “Uruguay” genovariant) associated with I. fuscipes ticks, expanding regional diversity and advancing understanding of the pathogen’s ecology and systematics. Moreover, the close phylogenetic proximity of the Uruguayan lineage to ecotypes V and III, together with ecological evidence, raise the possibility that small mammals, in addition to cervids like S. gouazoubira, may contribute to its maintenance, a hypothesis that requires confirmation. While these data suggest a potentially novel ecotype, formal recognition requires further evidence from a broader sampling of ticks and vertebrate hosts. Overall, our findings shed light on the potential involvement of I. fuscipes ticks in local A. phagocytophilum enzootic cycles and highlight the necessity for broader, integrated host–vector investigations across South America. However, until further comprehensive studies have addressed expanded tick and vertebrate host sampling, vector competence, host range, transmission dynamics and pathogenicity, the ecological and epidemiological implications of the A. phagocytophilum “Uruguay” lineage remains unresolved.
Supplementary Information
Additional file 1: Table S1. BLASTn matches of partial cytochrome* c* oxidase I (cox1) sequences retrieved from Ixodes fuscipes collected in Uruguay.Additional file 2: Table S2. BLASTn comparisons of Anaplasma phagocytophilum sequences for each locus (16S rRNA, gltA and groEL).Additional file 3: Table S3. GenBank accession numbers of Anaplasma spp. sequences used for phylogenic analyses based on 16S rRNA (rrs) and gltA genes. Sequences generated in this study are highlighted in bold.Additional file 4: Table S4. GenBank accession numbers of the Anaplasma spp. sequences used for phylogenic analyses based on groEL gene. Sequences generated in this study are highlighted in bold.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Culda CA, Panait LC, Cazan CD, Sprong H, Vinueza RL, Páez-Rosas D, et al. First report of Anaplasma phagocytophilum in Galapagos: high prevalence in dogs and circumstantial evidence for the role of Rhipicephalus linnaei as vector. Transbound Emerg Dis. 2025:5542334. 10.1155/tbed/5542334.10.1155/tbed/5542334 PMC 1224551440641950 · doi ↗ · pubmed ↗
- 2Chae JS, Foley JE, Dumler S, Madigan JE. Comparison of the nucleotide sequences of 16S r RNA, Ep-ank, and gro ESL heat shock operon genes in naturally occurring Ehrlichia equi and human granulocytic ehrlichiosis agent isolates from Northern California. J Clin Microbiol. 2000;38:1364–9. 10.1128/JCM.38.4.1364-1369.2000.10.1128/jcm.38.4.1364-1369.2000 PMC 8644610747108 · doi ↗ · pubmed ↗