Unraveling the molecular mechanisms in severe Alzheimer’s disease based on transcriptomic data using next generation knowledge discovery methods
Hind A. Alkhatabi, Alaa G. Alahmadi, Muhammad Imran Naseer, Peter Natesan Pushparaj

TL;DR
This study uses brain RNA data to uncover key molecular pathways involved in severe Alzheimer’s disease, focusing on neuroinflammation and energy metabolism.
Contribution
Novel application of next-generation knowledge discovery methods to identify dysregulated pathways in severe Alzheimer’s disease.
Findings
Dysregulation of energy metabolism and protein synthesis in severe Alzheimer’s disease.
Enrichment of neuroinflammatory pathways and immune system activation in affected brain regions.
Downregulation of ribosome biogenesis and oxidative phosphorylation in severe Alzheimer’s disease.
Abstract
Alzheimer’s disease (AD) is characterized by gradual cognitive decline. Here, we deciphered the molecular mechanisms using transcriptomic data derived from the dorsolateral prefrontal cortex (DLPFC) of patients with severe AD using next-generation knowledge discovery (NGKD) techniques. RNA sequencing data from the Gene Expression Omnibus (GEO) database (GSE53697) derived from the DLPFC of individuals with severe AD and healthy controls, obtained originally from frozen brain tissues of individuals classified based on the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria, and differentially expressed genes (DEGs) were identified by GEO2R analysis. The WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) was used for overrepresentation analysis (ORA) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database and Gene Set Enrichment Analysis (GSEA)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
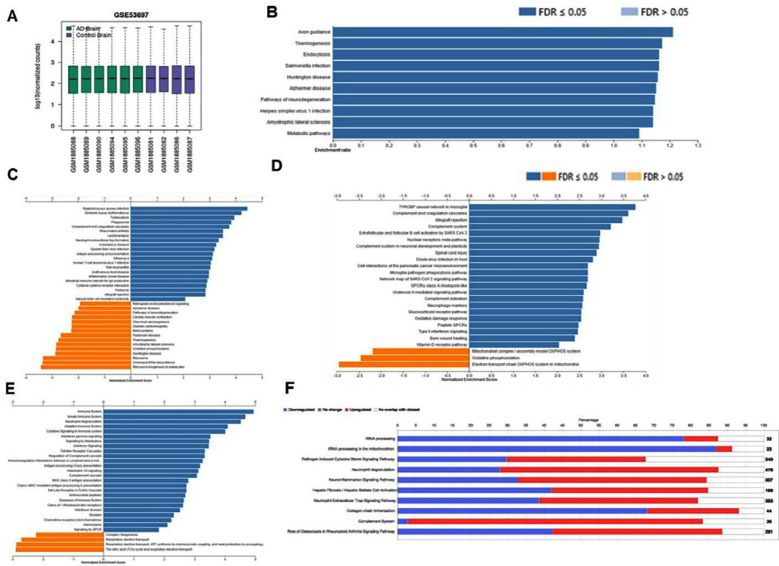 Fig.1
Fig.1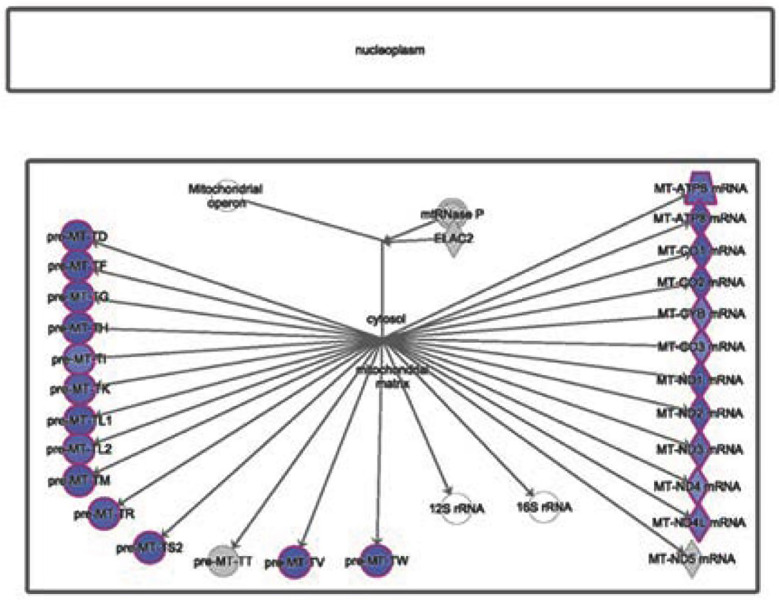 Fig.2
Fig.2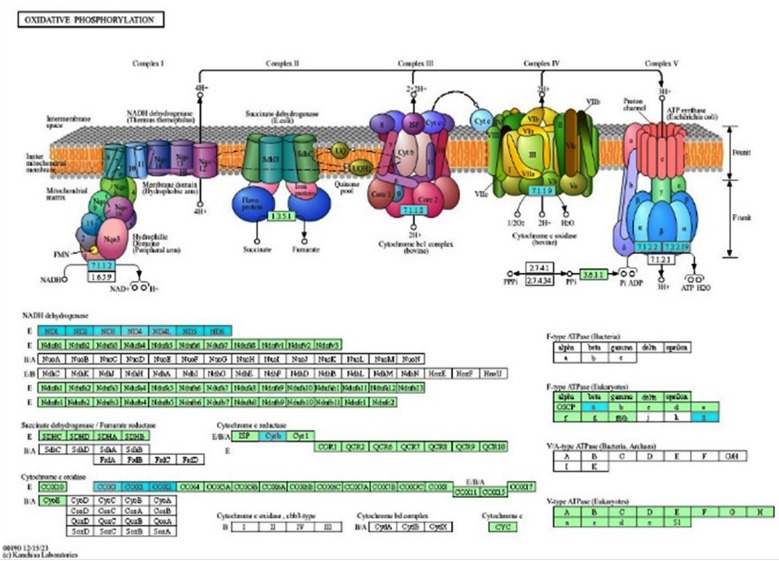 Fig.3
Fig.3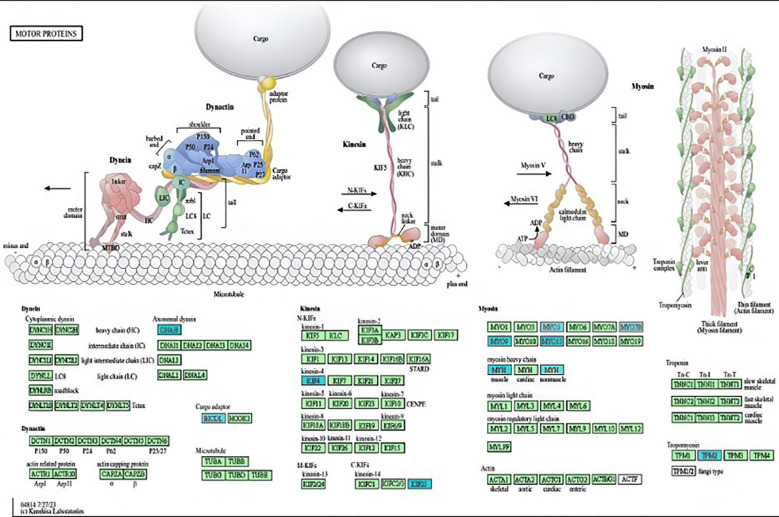 Fig.4
Fig.4| S. No. | Gene Name | Gene Symbol | Entrez Gene ID | Score |
|---|---|---|---|---|
| 1 | NADH dehydrogenase subunit 6 | ND6 | 4541 | -2.4743 |
| 2 | NADH dehydrogenase subunit 2 | ND2 | 4536 | -1.9249 |
| 3 | Cytochrome c oxidase subunit II | COX2 | 4513 | -1.5505 |
| 4 | NADH dehydrogenase subunit 1 | ND1 | 4535 | -1.5232 |
| 5 | NADH dehydrogenase subunit 5 | ND5 | 4540 | -1.5209 |
| 6 | Dynein axonemal heavy chain 1 | DNAH1 | 25981 | -1.5036 |
| 7 | Dynein axonemal heavy chain 2 | DNAH2 | 146754 | -1.4354 |
| 8 | Cytochrome c oxidase subunit I | COX1 | 4512 | -1.3766 |
| 9 | ATP synthase F0 subunit 8 | ATP8 | 4509 | -1.3395 |
| 10 | ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1 | ATP2A1 | 487 | -1.2187 |
| 11 | NADH dehydrogenase subunit 3 | ND3 | 4537 | -1.163 |
| 12 | ATP synthase F0 subunit 6 | ATP6 | 4508 | -1.1612 |
| 13 | NADH dehydrogenase subunit 4L | ND4L | 4539 | -1.1259 |
| 14 | Mitogen-activated protein kinase 12 | MAPK12 | 6300 | -1.0747 |
| 15 | Cytochrome b | CYTB | 4519 | -1.0096 |
| 16 | Cytochrome c oxidase subunit III | COX3 | 4514 | -0.9737 |
| 17 | NADH dehydrogenase subunit 4 | ND4 | 4538 | -0.9415 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAlzheimer's disease research and treatments · Bioinformatics and Genomic Networks · Neuroinflammation and Neurodegeneration Mechanisms
INTRODUCTION
Alzheimer’s disease (AD) is characterized by the gradual accumulation of extracellular β-amyloid (Aβ) plaques and intracellular neurofibrillary tangles composed of tau protein in the brain.1,2 Patients therefore exhibit progressive cognitive decline, memory loss, and behavioral changes.1–3 Typical features of cognitive loss in patients are memory impairment, aphasia, apraxia, agnosia, and disorientation.3,4 In addition, patients with AD may experience epileptic seizures, which complicate the clinical management of this debilitating disease.5 AD remains the fifth-leading cause of death among Americans aged 65 and older.3,5 Approximately 6.9 million Americans aged ≥65 years suffer from AD-associated dementia in the United States of America (USA).3,5 It was predicted that by 2060, 13.8 million individuals will be affected by AD in the absence of proper therapeutics to prevent, ameliorate, or cure AD. Globally, over 55 million individuals live with dementia, and the number of people with AD will double every two decades, potentially reaching approximately 78 million by 2030 and 139 million by 2050. An estimated 60–80 % of dementia cases are linked with AD and the majority of individuals experiencing brain changes and associated abnormalities due to a variety of etiologies of dementia.3,5
Due to difficulties in evaluating individuals with cognitive impairment6, the exact frequency of epileptic seizures in patients with AD remains unknown. Studies have reported glial activation, synaptic dysfunction, and marked neuronal loss in patients with severe AD.7-9 Oxidative stress, neuroinflammation, protein misfolding, and mitochondrial dysfunction are some of the characteristics of severe AD.10,11 Devising therapeutic modalities and improving treatment options depends on the understanding of molecular signaling pathways implicated in advanced or severe AD.
Recent findings based on next-generation knowledge discovery (NGKD) strategies have significantly expanded our knowledge and provided cues for further exploration of complex cellular and molecular mechanisms, enabling the identification of unique molecular markers, gene expression signatures, and protein-protein interactions associated with severe AD.3,12-14 As the exact mechanisms underlying the control of severe AD remain unknown, we investigated the complex molecular pathways associated with severe AD using NGKD tools. The dorsolateral prefrontal cortex (DLPFC) is vital for cognitive processes and memory.15 It was observed that the DLPFC displays structural atrophy like reduced cortical thickness and functional abnormalities, including diminished neuroplasticity and altered connectivity and are associated with cognitive impairments. 15Herein, we decipher the RNA sequencing (RNA-seq) data from the DLPFC of patients with severe AD using NGKD techniques.16
METHODOLOGY
We used RNA-seq data from the Gene Expression Omnibus (GEO) database (accession number: GSE53697) for this study. The dataset was deposited in GEO by Scheckel et al. (2016).15 Here, frozen brain tissues were obtained from the Mount Sinai Brain Bank of individuals classified based on the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria.15The DLPFC tissue was further processed, and RNA was extracted using the TRIzol method, followed by RNA-seq using an Illumina HiSeq 2500 (Illumina Corporation, USA).15 Although the study by Scheckel et al.15 (2016) included eight control subjects (without tangles or plaque pathology) and nine patients with advanced or severe AD (CDR 4-5), matched for age and sex, with brief postmortem intervals (PMI), we filtered and selected only four controls and six advanced AD samples (Fig.1A) for further downstream analysis using NGKD methods, as previously described.12-14
(A) RNA-seq data from the Gene Expression Omnibus (GEO) (accession no: GSE53697) were filtered, and only four controls and six severe AD samples were selected for further downstream analysis using next-generation knowledge discovery (NGKD) methods. (B) Overrepresentation analysis (ORA) of differentially expressed genes (DEGs) obtained from GEO2R analysis with a P-value ≤ 0.05, with Benjamini-Hochberg (BH) multiple testing correction based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. (C) Differentially regulated KEGG pathways, (D) Reactome pathways, (E) WikiPathways, and (F) canonical pathways based on Ingenuity Pathway Analysis (IPA) software core analysis in patients with severe AD.
Ethical statement:
Our study did not require ethical approval because it did not involve human or animal subjects and used existing RNA sequencing (RNA-seq) datasets from GEO. It was conducted entirely in a virtual environment and thus poses no risks to living subjects; therefore, Institutional Review Board (IRB) approval is not required for this study.3,13,14
GEO2R analysis:
The RNA-seq data from GSE53697 were analyzed with GEO2R using a robust multi-array average (RMA) algorithm to clean the background noise and ensure comparability between samples by reducing the technical variability and increasing the accuracy of downstream analyses using NGKD tools (access date: September 23, 2024). GEO2R was used to decipher differentially expressed genes (DEGs) between healthy controls and patients with severe AD using the statistical package “Linear Models for Microarray and RNA-Seq Data - limma” in R.14 For this purpose, linear models were fitted to the expression data for each gene by using empirical Bayes techniques to control for standard errors. Significant DEGs were selected using specific statistical thresholds such as P≤0.05 and the false discovery rate (FDR) using the Benjamini-Hochberg (BH) multiple testing correction method.
WebGestalt analysis:
An overrepresentation analysis (ORA) of DEGs in advanced AD compared to the control was performed using the WEb-based GEne SeT AnaLysis Toolkit (WebGestalt) (wGSEA) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Database with a P-value threshold of ≤ 0.05.17 As previously reported3,13, DEGs were further filtered using a fold-change criterion of ± 1.5 and analyzed by Gene Set Enrichment Analysis (GSEA) using WebGestalt (access date: September 23, 2024). Pathway analysis was performed using KEGG, Reactome, and Wikipathway databases. We selected Homo sapiens as the species, the gene symbol as ID, the genome as reference, and a minimum of five and a maximum of 2000 IDs (enrichment criteria) in each category, using the BH method with a significance level of P ≤ 0.05 to calculate the FDR.
Ingenuity pathway analysis:
Ingenuity Pathway Analysis (IPA) software (Qiagen, USA) was used to deduce the differentially regulated canonical pathways, upstream regulators, and non-directional gene networks in severe AD based on DEGs filtered with a cut-off of ± 1.5 and a P-value of less than 0.05. 13,18 Fisher’s exact test was used in IPA to calculate the Z-scores. Differentially regulated canonical pathways, upstream regulators, and undirected gene networks were visualized using IPA to further deduce AD-associated disease mechanisms.
RESULTS
GEO2R analysis of the RNA sequencing data derived from the DLPFC region of the brains of patients with severe AD and the normal control regions (Fig.1A) provided 24207 DEGs based on a P-value cut-off of ≤ 0.05, with BH multiple testing correction for FDR. Furthermore, this list was filtered based on a fold change of ± 1.5 to obtain 3103 DEGs for downstream processing and knowledge discovery. We used the WebGestalt tool to initially analyze the 24207 DEGs based on the Over Representation Analysis (ORA) method to evaluate the impact of advanced AD on the molecular mechanisms based on the KEGG pathway database (Fig.1B) and found that the pathways of neurodegeneration, Huntington’s disease, endocytosis, axon guidance, amyotrophic lateral sclerosis (ALS), Salmonella Infection, and metabolic pathways were significantly enriched (P ≤ 0.05) based on FDR in patients with AD.
Next, we used the GSEA method in WebGestalt to evaluate the DEGs filtered based on fold change to obtain differentially regulated KEGG, Reactome, and WikiPathways in patients with AD. Based on this analysis, we found that KEGG pathways such as AD, aminoacyl t-RNA biosynthesis, oxidative phosphorylation, motor proteins, amyotrophic lateral sclerosis (ALS), neurodegeneration, ribosome biogenesis in eukaryotes, and retrograde endocannabinoid signaling were significantly downregulated (P ≤ 0.05) in the AD group compared to the control group. In contrast, the DEGs implicated in neutrophil extracellular trap formation, Epstein-Barr virus infection, Staphylococcus aureus infection, antigen processing, and presentation were significantly (P ≤ 0.05) enriched in the AD group than in the control group (Fig.1C).
Reactome pathways, such as ATP synthesis by chemiosmotic coupling, Complex I biogenesis, respiratory electron transport, heat production by uncoupling proteins, and the citric acid (TCA) cycle, were negatively enriched (P ≤ 0.05) in patients with AD compared with the control group. Conversely, the Reactome pathways, such as GPCR signaling, infectious disease, immune system, cytokine, and chemokine signaling, were positively (P ≤ 0.05) enriched in the DLPFC of patients with AD compared to that of the control group (Fig.1D).
The electron transport chain oxidative phosphorylation (OXPHOS) system in mitochondria and mitochondrial complex I assembly model OXPHOS system based on WiKiPathways were negatively enriched (P ≤ 0.05) in patients with AD compared to controls. In contrast, WikiPathways, such as the complement system in neuronal development and plasticity, oxidative damage response, Type-II interferon signaling, tyrosine protein tyrosine kinase-binding protein (TYROBP) causal network in microglia, complement and coagulation cascades, Ebola virus infection in the host, and microglial pathogen phagocytosis pathway, were positively (P ≤ 0.05) enriched in patients with AD compared to the control group (Fig.1E). The canonical pathways from the IPA core analysis revealed that ribosomal RNA (rRNA) processing, transfer RNA (tRNA) processing in the mitochondria, and collagen chain trimerization were downregulated (Z-score ≤ -2). In contrast, canonical pathways, such as the pathogen-induced cytokine storm signaling pathway, neutrophil degranulation, neuroinflammation signaling pathway, neutrophil extracellular trap signaling pathway, and complement system, were enriched (Z score ≥ 2) in patients with severe AD compared to the control group (Fig.1F).
IPA analysis showed that genes encoding enzymes involved in rRNA processing, such as cytochrome c oxidase subunit I, cytochrome c oxidase subunit III, NADH dehydrogenase subunit 1, ATP synthase F0 subunit 8, NADH dehydrogenase subunit 2, NADH dehydrogenase subunit 3, ATP synthase F0 subunit 6, and cytochrome c oxidase subunit II (Fig.2), were negatively regulated in the severe AD group.
Ribosomal RNA (rRNA) RNA-processing pathway in AD. The downregulated genes involved in the rRNA processing canonical pathway were obtained from IPA software (based on negative Z-scores, highlighted in blue) in severe AD.
The genes involved in oxidative phosphorylation (highlighted in turquoise), such as NADH dehydrogenase subunit 2, ATP synthase F0 subunit 8, NADH dehydrogenase subunit 6, ATPase H+/K+ transporting subunit alpha, cytochrome c oxidase subunit I, cytochrome c oxidase subunit II, ATP synthase F0 subunit 6, and NADH dehydrogenase subunit 4, based on KEGG pathway database were significantly downregulated (P ≤ 0.05) in patients with AD (Fig.3).
Oxidative phosphorylation pathway based on the KEGG database in severe AD. The downregulated genes involved in oxidative phosphorylation (highlighted in turquoise) in severe AD were identified using the GSEA method in WebGestalt.
Moreover, KEGG pathway analysis showed that genes coding for motor proteins (highlighted in turquoise), such as dynein axonemal heavy chain 1, myosin heavy chain 3, kinesin family member 25, myosin IXB, kinesin family member 4 B, myosin XVB, tropomyosin 2, and BICD family members, such as cargo adaptor 1, dynein axonemal heavy chain 2, myosin heavy chain 7 B, myosin VIIB, myosin VC, and myosin heavy chain 14, were significantly (P ≤ 0.05) downregulated in AD patients (Fig.4).
Motor protein pathways based on the KEGG database in patients with severe AD. Downregulated genes involved in the motor protein pathway (highlighted in turquoise) in severe AD were derived using the GSEA method in WebGestalt.
Interestingly, an array of significantly downregulated genes (P ≤ 0.05) involved in neurodegeneration pathways (Table-I), including dynein axonemal heavy chain 1, NADH dehydrogenase subunit 6, mitogen-activated protein kinase 12, NADH dehydrogenase subunit 1, ATPase sarcoplasmic/endoplasmic NADH dehydrogenase subunit 3, cytochrome c oxidase subunit III, NADH dehydrogenase subunit 4, cytochrome c oxidase subunit I, reticulum Ca2+ transporting 1, cytochrome b, NADH dehydrogenase subunit 5, dynein axonemal heavy chain 2, and NADH dehydrogenase subunit 4 L, were also involved in AD signaling and OXPHOS in AD patients.
DISCUSSION
AD is characterized by a slow loss of cognitive abilities, memory, and behavior that affects daily life and quality of life (QoL).1,2,3,12 In the present study, we observed marked changes in gene expression potentially regulating an array of molecular signaling pathways in the DLPFC of patients with severe AD compared to healthy controls. Here, the NGKD analysis of DEGs derived from patients with severe AD, based on both open-source tools such as WebGestalt and IPA, a commercial software, provided pivotal evidence of the differently regulated complex molecular landscape in severe AD.
Our NGKD analysis revealed the dysregulation of axon guidance, endocytosis, and various neurodegenerative processes in severe AD, as observed in previous studies. 3,18,19 The dysregulation of axon guidance leads to abnormal neuronal connectivity and impaired endocytosis which can significantly impair amyloid-beta clearance, thus increasing the accumulation of pathological aggregates in the AD brain.20–22 AD dysregulates retrograde endocannabinoid signaling, motor proteins, and oxidative phosphorylation. Synaptic plasticity, neuroprotection, and inflammation control are dependent on these factors. Disruption of this signaling pathway in AD can lead to progressive loss of neurons and cognitive impairment. 23 Cognitive decline in AD has been linked to dysfunction of the endocannabinoid system. Proper axonal transport is essential for maintaining ideal neuronal health and synaptic functions; hence, the dysregulation of motor proteins involved in axonal transport contributes to synaptic dysfunction and neuronal loss in AD.24,25 More importantly, the dysregulation of motor protein genes, as observed in our study in severe AD, can lead to the accumulation of harmful proteins and organelles, causing a slow loss of neurons in the brain. In addition, the NADH dehydrogenase subunits and cytochrome c oxidase subunits involved in the electron transport chain are downregulated in AD, which could significantly impair energy metabolism in the neurons of patients with AD. Mitochondrial dysfunction may substantially reduce ATP production, increase oxidative stress, and impair calcium homeostasis, thereby resulting in neuronal death in patients with AD, as previously described.3 Antigen processing and presentation, neutrophil extracellular trap (NET) formation, and cytokine signaling are upregulated in AD. As observed in our study, the immune activation in the brain could result in the release of pro-inflammatory mediators, increased oxidative stress, and neurotoxicity, further exacerbating neuronal damage or loss of neurons and resulting in progressive cognitive decline.26-28
The immune responses against pathogens such as Staphylococcus aureus and Epstein-Barr virus may further augment the chronic inflammatory state observed in AD brains, as we observed that the molecular pathways related to these microbes were positively enriched in the DLPFC of patients with severe AD.29 Ribosome biogenesis plays an important role in normal neuronal function30,31, and the accumulation of tau proteins and β-amyloid (Aβ) (1-42) causes progressive neuronal loss, the formation of neuronal fiber tangles (NFTs), and senile plaque (SP) deposition in neurons.32 Studies have demonstrated that pathological tau proteins are abnormally associated with ribosomes in AD brains, leading to impaired protein synthesis.33 This aberrant tau-ribosome interaction significantly reduces the production of nascent proteins that are important for synaptic plasticity and are critical for learning and memory. 30, 31, 34 Ribosomal dysfunction that leads to impaired translation has been associated with memory loss in tauopathies such as AD.34
Furthermore, studies have demonstrated that tau oligomerization induces translational stress response and inhibits protein synthesis. An array of ribosomal proteins was identified based on mass spectrometry that selectively bind to oligomeric tau (oTau), thereby affecting quality control mechanisms related to ribosomal biogenesis and RNA translation.35 These findings provides a mechanistic link between the presence of pathological tau within brain cells and cognitive impairments evident in virtually all tauopathies, including AD.22,31,34 Efficient axonal transport is essential for the delivery of newly synthesized proteins, lipids, and organelles to synapses, and for the removal of damaged components. The defective axonal transport mechanisms in AD patients could be observed due to synaptic dysfunction, axonal swelling, and neuronal death. The dysregulation of ribosome biogenesis and protein synthesis, association of pathological tau proteins with ribosomes impairs protein synthesis, reduces the production of proteins vital for synaptic plasticity may potentially lead to cognitive decline in AD patients. Dysregulated protein synthesis affects cellular stress responses and proteostasis, which adversely affects neuronal health.33-36
Strength of the Study:
The main strength of our study is that we generated a comprehensive NGKD analysis of RNA-seq data from DLPFC tissues from patients with severe AD, and our approach is highly effective for analyzing high-throughput expression data to decode differentially regulated molecular pathways related to severe AD. In addition, our study is particularly helpful for decoding complex diseases, such as AD, in which the interaction of an array of pathological mechanisms is responsible for the entire disease state. The key DEGs deduced from our study may be further explored in detail using appropriate in vitro and in vivo systems and the latest artificial intelligence/machine learning classification algorithms to identify biomarker signatures associated with the diagnosis, staging, or monitoring of AD progression.
Limitations
However, our study has some limitations due to the small number of samples in the AD group and reliance on only high-dimensional datasets derived from RNA sequencing. Hence, the identification of molecular pathways using RNA-seq and other “omics” datasets and extensive NGKD analysis is essential to develop and design experimental strategies for personalized treatment regimens for patients with AD.
CONCLUSIONS
Our study unraveled the complex molecular pathways involved in AD pathogenesis. This includes the disruption of energy metabolism, protein synthesis, axonal transport, and immunological responses. Importantly, we obtained additional insights into the cascade of molecular pathways leading to severe AD and identified important avenues for therapeutic intervention. Our findings require further validation using appropriate model systems and should concentrate mainly on their potential for developing novel diagnostic tools and therapeutic strategies for AD. In the future, the functional effects of the dysregulated pathways identified in this study should be justified using appropriate in vitro models, such as induced pluripotent stem cell (iPSC)-derived neurons or glial co-cultures, and in vivo AD models. As stated in our limitations, these analyses should be performed in larger and more diverse AD patient cohorts. Finally, integrating proteomics, glycomics, metabolomics, and epigenomics is necessary to construct a more complete model of AD and to fully understand the sequence of events that lead from molecular dysregulation to severe cognitive decline. In conclusion, our findings add to the existing knowledge of the pathophysiology of severe AD and will help in the development of more effective strategies for diagnosis, therapy, and prevention.
Authors’ contribution:
HK & PNP conceived, designed, writing original draft, review and editing; are responsible for integrity of research.
HK, AA, MIN & PNP did data collection, data curation, data analysis and have read and agreed to the final version of the manuscript.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Scheltens P De Strooper B Kivipelto M Holstege H Chételat G Teunissen CE Alzheimer's disease Lancet.20213971028415771590 doi:10.1016/S 0140-6736(20)32205-43366741610.1016/S 0140-6736(20)32205-4PMC 8354300 · doi ↗ · pubmed ↗
- 2Knopman DS Amieva H Petersen RC Chételat G Holtzman DM Hyman BT Alzheimer disease Nat Rev Dis Primers.20217133 doi:10.1038/s 41572-021-00269-y 3398630110.1038/s 41572-021-00269-y PMC 8574196 · doi ↗ · pubmed ↗
- 3Alkhatabi HA Pushparaj PN Untangling the complex mechanisms associated with Alzheimer's disease in elderly patients using high-throughput RNA sequencing data and next-generation knowledge discovery methods:Focus on potential gene signatures and drugs for dementia Heliyon.2024111 e 41266 doi:10.1016/j.heliyon.2024.e 412663983444010.1016/j.heliyon.2024.e 41266 PMC 11743088 · doi ↗ · pubmed ↗
- 4Zverova M Clinical aspects of Alzheimer's disease Clin Biochem.20197236 doi:10.1016/j.clinbiochem.2019.04.0153103480210.1016/j.clinbiochem.2019.04.015 · doi ↗ · pubmed ↗
- 52024 Alzheimer's disease facts and figures Alzheimers Dement 202420537083821 doi:10.1002/alz.138093868939810.1002/alz.13809 PMC 11095490 · doi ↗ · pubmed ↗
- 6Yang F Chen L Yu Y Xu T Chen L Yang W Alzheimer's disease and epilepsy:An increasingly recognized comorbidity Front Aging Neurosci.t 202214940515 doi:10.3389/fnagi.2022.94051510.3389/fnagi.2022.940515 PMC 968517236438002 · doi ↗ · pubmed ↗
- 7Novak A Vizjak K Rakusa M Cognitive Impairment in People with Epilepsy J Clin Med.202211(1)267doi:10.3390/jcm 110102673501200710.3390/jcm 11010267 PMC 8746065 · doi ↗ · pubmed ↗
- 8Li J Wu X Tan X Wang S Qu R Wu X The efficacy and safety of anti-Aβagents for delaying cognitive decline in Alzheimer's disease:a meta-analysis Front Aging Neurosci.2023151257973 doi:10.3389/fnagi.2023.12579733802076310.3389/fnagi.2023.1257973 PMC 10661413 · doi ↗ · pubmed ↗