Coronary artery spasm: mechanisms, risk factors, and translational strategies for precision management
Zhihui Kuang, Ranran Kong, Zhonghua Wang, Siying Zuo, Liwei You, Xuan Wang, Xiaoyun Si, Jinfeng Liang

TL;DR
This review explores coronary artery spasm, its mechanisms, risk factors, and strategies for better diagnosis and treatment.
Contribution
The paper integrates mechanisms and clinical insights to propose precision management strategies for coronary artery spasm.
Findings
CAS is often underrecognized due to limited availability of pharmacological provocation testing.
CAS is linked to smoking, inflammation, and genetic factors, but not consistently to hypertension or diabetes.
Microvascular spasm and endothelial dysfunction are key mechanisms in CAS.
Abstract
Coronary artery spasm (CAS) is a major form of coronary vasomotor dysfunction within ischemia with non-obstructive coronary arteries (INOCA). Despite ethnic and geographic variation in prevalence, CAS is underrecognized because confirmation often requires pharmacological provocation testing that is unavailable in many centers. Clinically, CAS ranges from silent ischemia and angina to acute myocardial infarction, malignant arrhythmias, heart failure, and sudden cardiac death. Evidence suggests that spasm often occurs at sites with mild atherosclerosis and that its risk profile differs from atherosclerotic cardiovascular disease, with more consistent links to smoking, inflammatory burden, and genetic susceptibility, while associations with hypertension and diabetes remain inconsistent. Advances in invasive coronary function testing and recognition of microvascular spasm support an…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
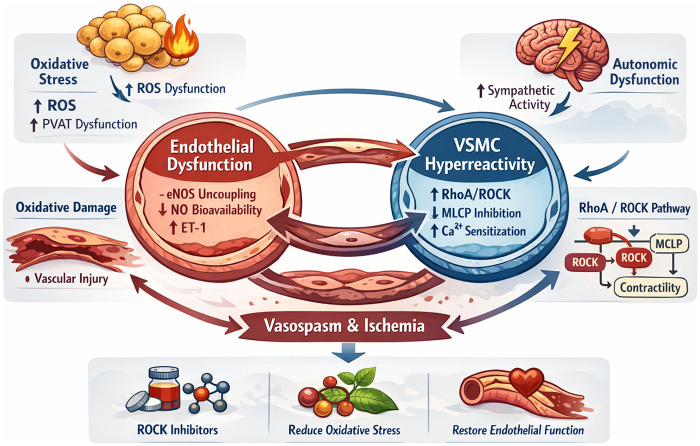 Figure 1
Figure 1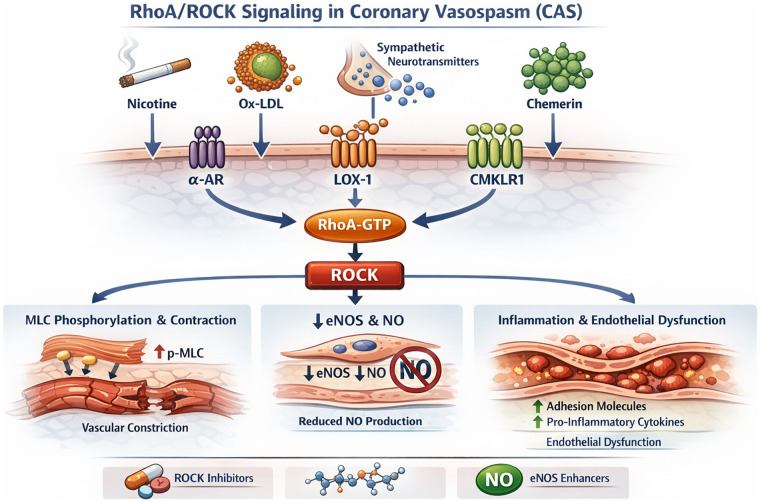 Figure 2
Figure 2| Risk factor category | Specific factor | Core mechanism | Epidemiological evidence | Evidence level | Clinical recommendation | Recommendation strength/notes |
|---|---|---|---|---|---|---|
| Modifiable Lifestyle Factors | Smoking | Smoking induces endothelial dysfunction, increases oxidative stress, and activates the RhoA/ROCK signaling pathway, thereby enhancing vascular smooth muscle contractility. | Meta-analysis ( | High (observational + guideline-endorsed risk factor) | Complete smoking cessation; integrate into comprehensive cardiac rehabilitation | Guideline-recommended smoking cessation for INOCA risk management (Class I, LOE A) ( |
| Modifiable Lifestyle Factors | Dyslipidemia/Atherosclerotic burden | ox-LDL–related endothelial injury, inflammation, ROCK activation, increased VSMCs contractile sensitivity | Cross-sectional study ( | Moderate (observational; heterogeneous) | Manage lipids per general dyslipidemia guidelines; intensify LDL-C lowering in high/very-high CV risk | ESC/EAS LDL-C targets (<1.8 mmol/L for high risk; <1.4 mmol/L for very-high risk) rather than CAS-specific targets ( |
| Environmental/Drug Factors | Airborne particulate matter (PM2.5/PM10) | Systemic inflammation, endothelin-1/ROCK activation, endothelial dysfunction | Observational study ( | Low–Moderate (observational) | Exposure reduction strategies (avoid high-PM days; respirator masks for high-risk) | Pragmatic risk reduction; not CAS-specific guideline class—state as preventive advice based on observational evidence |
| Environmental/Drug Factors | Vasoconstrictive drugs (e.g., cocaine; nonselective β-blockers; calcineurin inhibitors) | Case report: local anesthetic cocaine induced severe CAS ( | Low (case-based/experimental) | Avoid cocaine and triggers; avoid nonselective β-blockers in suspected/known VSA; monitor high-risk exposures | JCS guidance notes concern β-blockers may exacerbate spasm; use caution in high-risk CAS contexts ( | |
| Anatomical/Comorbid Conditions | MB | Coronary flow turbulence and vessel compression contribute to endothelial dysfunction and abnormal vasomotor regulation. | Cohort ( | Moderate (observational) | Symptom-guided management; consider β1-selective blockers for MB-related exertional symptoms | Important nuance: β-blockers can worsen VSA; if CAS coexists, prioritize CCBs/nitrates and individualizeβ1-selective use |
| Anatomical/Comorbid Conditions | OSA | Intermittent hypoxia leads to sympathetic activation, oxidative stress, and endothelial dysfunction. | Case–control ( | Low for CAS-specific; High for CV risk | Screen suspected OSA; treat per sleep medicine standards (CPAP when indicated) | CPAP improves overall CV risk and endothelial function, CAS-specific outcomes remain unproven |
| Anatomical/Comorbid Conditions | Thyrotoxicosis | Excess thyroid hormone increases myocardial oxygen demand, reduces coronary vasodilator reserve, and heightens catecholamine sensitivity. | Retrospective cohort ( | Low–Moderate (observational) | Prompt thyroid normalization; avoid triggers during thyrotoxic phase | General medical management; not CAS-specific |
| Non-Modifiable Factors | Age | Endothelial senescence, oxidative stress, reduced NO bioavailability | Retrospective observational study ( | High (large observational cohorts) | Consider provocation testing based on symptoms/INOCA context rather than age alone | JCS provides indications for provocation testing; avoid presenting age as stand-alone indication (use symptom-driven testing) ( |
| Non-Modifiable Factors | Sex | Men: more epicardial spasm (often higher smoking exposure); women: microvascular spasm enriched, post-menopause risk | Gender–stratified analysis ( | Moderate (observational) | Phenotype-specific evaluation (epicardial vs. microvascular) and risk factor control | Highlight diagnostic heterogeneity |
| Genetic Variants | ALDH2*2, eNOS Glu298Asp, RNF213, etc. | Aldehyde detox/NO signaling/vascular cell survival pathways | Genomic studies identified ALDH2*2 frequency as markedly higher in East Asians (∼30%–40%) ( | Moderate–High (genetic association) | Not routine screening; consider in strong family history/sudden death or refractory phenotypes | Present as risk modifiers, not direct clinical test recommendations unless local practice supports |
| Controversial/Inconsistent Factors | Alcohol | In ALDH2*2 carriers: acetaldehyde accumulation, prostanoid imbalance; autonomic effects | Korean cohort ( | Low–Moderate (observational; confounded) | Avoid heavy drinking; consider stricter avoidance in ALDH2*2 carriers | Lifestyle advice; causality uncertain |
| Controversial Factors | Hyperuricemia | eNOS inhibition, endothelin/ROCK activation | Cohort ( | Low–Moderate (observational) | No routine urate-lowering solely for CAS; manage per gout/CKD indications | Marker hypothesis; evidence inconsistent |
| Controversial Factors | Hypertension | Vascular remodeling; endothelial dysfunction | Cohort ( | Low–Moderate (heterogeneous observational) | Standard blood pressure control per guidelines | No CAS-specific recommendation |
| Controversial Factors | Glucose metabolism disorders/Diabetes | Fibrosis/endothelial dysfunction; treatment confounding | Cohort ( | Low–Moderate (heterogeneous observational) | Standard glycemic control; assess coronary microvascular dysfunction (CMD) when clinically suspected. | Relevance mainly for microvascular dysfunction |
| Gene variant | Ethnic distribution | Core mechanism | CAS risk | Evidence type | Key references |
|---|---|---|---|---|---|
| ALDH2*2 | Predominantly East Asian; high carrier frequency in East Asia; impacts alcohol flushing phenotype | Reduced aldehyde dehydrogenase-2 enzymatic activity leads to accumulation of reactive aldehydes, increased oxidative stress, and endothelial dysfunction. | Reported OR 3.00 (95% CI: 1.90–4.80) | Candidate-gene case–control association | JCS VSA guideline ( |
| eNOS Glu298Asp | Worldwide distribution. | Reduced endothelial nitric oxide synthase stability and activity, leading to decreased NO bioavailability. | Reported OR 2.83 (95% CI: 1.25–6.41) | Candidate-gene case–control association | Chang et al. ( |
| RNF213 p.R4810K | Predominantly East Asian; low carrier rate. | Variant-associated endothelial and vascular smooth muscle cells dysfunction with increased susceptibility to apoptosis. | Reported OR 2.34 (95% CI: 1.99–2.74) | Population-specific variant association (East Asian cohorts) | Hikino et al. ( |
| EDN1 rs5370 | Multi-ethnic distribution. | Enhanced endothelin-1 gene expression and vasoconstrictor signaling. | Reported OR 1.75 ( | Candidate-gene case–control association | Lee et al. ( |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiac Imaging and Diagnostics · Cardiovascular Health and Disease Prevention · Coronary Interventions and Diagnostics
Introduction
1
Coronary artery spasm (CAS) is a major form of coronary vasomotor dysfunction and a key endotype within ischemia with non-obstructive coronary arteries (INOCA). Although prevalence varies across ethnic and geographic populations, CAS remains underrecognized because the diagnosis often relies on pharmacological provocation testing that is not routinely performed in many centers. Clinically, CAS ranges from silent myocardial ischemia and angina to acute myocardial infarction, life-threatening ventricular arrhythmias, heart failure, and sudden cardiac death (1).
Early pathological and intravascular imaging studies suggest that CAS frequently occurs at sites with subclinical or mild atherosclerosis, indicating a functional–structural interplay between vasomotor instability and atherosclerotic remodeling (2). Observational comparisons further indicate that the risk factor profile of CAS differs from that of atherosclerotic cardiovascular disease (ASCVD): associations with hypertension and diabetes are inconsistent or weak, whereas smoking, inflammatory burden, and genetic susceptibility show more reproducible links and stronger mechanistic plausibility (3, 4).
In parallel with mechanistic heterogeneity, CAS also exhibits substantial clinical phenotype heterogeneity. CAS can present across distinct ischemic phenotypes, including angina with non-obstructive coronary arteries (ANOCA) and myocardial infarction with non-obstructive coronary arteries (MINOCA). Emerging outcome data suggest worse prognosis in CAS-related MINOCA than in CAS presenting as ANOCA (5), underscoring the need for phenotype-informed risk stratification and management. These observations align with recent calls for precision approaches in vasomotor disorders (6), emphasizing that CAS should not be viewed as a uniform clinical entity.
Systematic synthesis of the risk factors and pathobiological pathways underlying CAS is essential for advancing precision prevention, individualized diagnosis, and targeted therapeutic strategies. To improve interpretability and avoid overstatement, we apply a structured evidence framework throughout this review. We first summarize the pathophysiological mechanisms of CAS, then integrate its risk factor landscape, and finally discuss translational implications and future research priorities.
Methodological approach of this review
2
To enhance interpretability and avoid overstatement, we apply a structured evidence framework throughout this review. We distinguish observational clinical studies (cohort, case–control, registry, and case-series data), mechanistic and experimental investigations (cellular, animal, and human physiological studies), and interventional clinical evidence (randomized or non-randomized therapeutic studies). We describe observational findings as associations, mechanistic studies are framed as biological plausibility, and causal language is reserved for interventional evidence or guideline-endorsed recommendations. Where applicable, guideline statements are reported using Class of Recommendation (COR) and Level of Evidence (LOE).
Microvascular spasm: diagnostic limitations and the epicardial–microvascular continuum
3
Microvascular spasm is increasingly recognized in vasomotor dysfunction, yet its diagnosis is less straightforward than epicardial spasm because it lacks a direct angiographic correlate. Current definitions rely on acetylcholine-provoked ischemic symptoms and/or ischemic ECG changes without angiographic epicardial constriction (consensus criteria) (7, 8). As a result, diagnostic classification can be sensitive to protocol differences (e.g., acetylcholine dosing), interpretive thresholds, and test–retest/inter-operator variability in symptom and ECG assessment, motivating ongoing standardization of invasive testing (9, 10). Mechanistic and treatment data for microvascular spasm remain largely derived from small physiological or observational studies, whereas epicardial spasm is supported by larger provocation-tested cohorts and more extensive clinical evidence (8, 11, 12).
Emerging invasive data and contemporary syntheses suggest that epicardial and microvascular spasm often overlap and may represent a continuum of coronary vasomotor dysregulation driven by shared upstream pathways [e.g., endothelial dysfunction, Rho-kinase (ROCK) signaling, autonomic imbalance, inflammation] (13, 14). This framing helps interpret heterogeneous results and prevents overgeneralization of therapies across distinct spasm endotypes (10, 13, 14).
CAS can present as ANOCA or MINOCA, and this distinction has pragmatic implications for risk stratification and follow-up. In ANOCA, CAS is often managed as a recurrent symptom disorder, with emphasis on trigger identification, optimization of vasodilator therapy, and relapse prevention, while follow-up is largely guided by symptom control and functional limitation. In MINOCA, spasm-positive testing occurs within an acute coronary syndrome phenotype, where short-to-intermediate event risk and recurrent ischemic injury become more central; closer surveillance and a lower threshold for escalation of preventive strategies may therefore be appropriate (5). Because MINOCA is mechanistically heterogeneous, a spasm finding should be interpreted alongside evaluation for competing or concomitant mechanisms (e.g., plaque disruption or thromboembolism) where clinically feasible, rather than treated as a sole explanation. This phenotype-informed framing aligns with emerging precision approaches in vasomotor disorders (6).
Pathophysiological mechanisms: the central pathobiological network—an “Amplification Loop” linking endothelial dysfunction to smooth muscle hyperreactivity
4
CAS is best understood as a dynamic, self-reinforcing network rather than a linear sequence of events (Figure 1). This network is anchored by endothelial dysfunction and vascular smooth muscle cell (VSMC) hyperreactivity and is further shaped by inflammation–oxidative stress coupling, autonomic dysregulation, and genetic susceptibility. Perturbation at any node may propagate across the system and lower the threshold for spasm.
Pathological amplification loop in CAS. This figure illustrates the interconnected molecular and physiological processes that form a self-reinforcing pathological amplification loop in CAS. The loop is centered on endothelial dysfunction and vascular smooth muscle cell (VSMC) hyperreactivity, integrating inflammatory, oxidative, autonomic, and genetic mechanisms. Endothelial injury characterized by eNOS uncoupling, reduced nitric oxide (NO) bioavailability, and increased endothelin-1 (ET-1) expression initiates vasomotor instability. Inflammatory activation and oxidative stress promote perivascular adipose tissue (PVAT) dysfunction, excessive reactive oxygen species (ROS) generation, and propagation of vascular injury. Activation of the RhoA/ROCK signaling cascade enhances VSMC contractility through myosin light chain phosphatase (MLCP) inhibition and calcium sensitization, while autonomic dysregulation provides physiological triggers for spasm onset. Sustained vasospasm induces ischemia, which further aggravates endothelial injury and oxidative stress, thereby maintaining the vicious cycle. This integrated network highlights potential therapeutic targets, including ROCK inhibition, oxidative stress reduction, and restoration of endothelial function, which may disrupt the amplification loop and prevent recurrent spasm. eNOS, endothelial nitric oxide synthase; ROS, reactive oxygen species; ROCK, Rho-associated coiled-coil containing protein kinase; PVAT, perivascular adipose tissue; VSMC, vascular smooth muscle cell; ET-1, endothelin-1; MLCP, myosin light chain phosphatase; NO, nitric oxide.
Dual core pathways: coordinated activation of endothelial dysfunction and VSMCs hyperreactivity
4.1
Endothelial dysfunction is a core component of CAS pathophysiology, driven by reduced nitric oxide (NO) bioavailability and an imbalance between vasodilator and vasoconstrictor influences. Reduced endothelial nitric oxide synthase (eNOS) activity and tetrahydrobiopterin (BH4) can promote eNOS uncoupling, diminishing NO production and increasing superoxide generation. In parallel, elevated vasoconstrictors such as endothelin-1 (ET-1), serotonin, and histamine may further bias vascular tone toward constriction (15).
VSMCs hyperreactivity serves as a major downstream effector of CAS. RhoA/ROCK signaling is strongly implicated, increasing Ca²^+^ sensitization by inhibiting myosin light-chain phosphatase (MLCP) and thereby facilitating myosin light-chain phosphorylation and sustained contraction. Impaired KATP channel function and increased T-type calcium channel activity may further contribute to hyperreactivity in experimental and clinical physiological studies (16). Together, endothelial dysfunction and VSMCs hyperreactivity create a permissive substrate for coronary spasm.
Central amplifier: the inflammation–oxidative stress– perivascular adipose tissue (PVAT) axis
4.2
Inflammatory activation can bridge endothelial dysfunction and VSMCs hyperreactivity. Inflammatory mediators such as high-sensitivity C-reactive protein (hs-CRP) and interleukin-6 (IL-6) have been associated with endothelial injury, ROS generation, and RhoA/ROCK activation in mechanistic and translational studies (17). Oxidative stress can further deplete NO bioavailability, promote eNOS uncoupling, and facilitate oxidation of low-density lipoprotein cholesterol (LDL-C) to oxidized LDL (ox-LDL), thereby sustaining an inflammation–oxidative stress feedback loop that may exacerbate endothelial dysfunction and vascular remodeling (18).
Perivascular Adipose Tissue (PVAT) has also emerged as an active paracrine organ that may modulate vascular homeostasis. Under physiological conditions, PVAT exerts anti-contractile effects through adiponectin-related signaling and anti-inflammatory mediators (19). In obesity and related metabolic states, PVAT may shift toward a pro-contractile phenotype, with increased release of mediators such as chemerin, endothelin-1, TNF-α, and oxidative enzymes, alongside reduced adiponectin. PVAT-derived chemerin has been linked to enhanced VSMCs contractility via RhoA/ROCK signaling and to oxidative stress–related endothelial dysfunction (20), whereas reduced adiponectin may weaken vasodilatory and anti-proliferative buffering (21). This shift may contribute to a local milieu favoring vasoconstriction and impaired endothelial reserve (19, 22).
Maging and histopathological studies further suggest associations between PVAT phenotype and vascular reactivity. Quantitative coronary computed tomography angiography (CCTA) analyses report that the PVAT attenuation index correlates with the incidence and severity of vasospasm (23), suggesting that PVAT may act as a metabolic–inflammatory intermediary linking systemic status to local vasomotor instabilit (22).
Triggering regulatory axis: bidirectional modulation by the autonomic nervous system and circadian rhythmicity
4.3
The autonomic nervous system (ANS) is implicated in CAS, with sympathetic overactivity contributing to coronary spasm. Sympathetic activation via norepinephrine stimulates α-adrenergic receptors on VSMCs, increases intracellular Ca²^+^, and activates RhoA/ROCK signaling, thereby enhancing VSMCs contractility (24, 25).
Under physiological conditions, parasympathetic activation via acetylcholine (ACh) induces endothelium-dependent vasodilation through NO release. In the setting of endothelial dysfunction, however, ACh may paradoxically evoke direct smooth muscle constriction and precipitate spasm. Seminal clinical investigations have shown that intracoronary ACh infusion can reliably provoke spasm in patients with variant angina—a response abolished by atropine—supporting a pathological shift of parasympathetic signaling from vasodilatory to vasoconstrictive dominance (25, 26).
Evidence from ischemic heart disease suggests that vagus nerve stimulation (VNS), via invasive or transcutaneous approaches, may rebalance autonomic tone and modulate inflammatory signaling; however, its efficacy and safety for CAS remain unproven and should be framed as a hypothesis requiring CAS-specific clinical outcome studies (27).
Clinical autonomic monitoring studies have described dynamic pre-spasm shifts. Heart rate variability analyses indicate that, within minutes preceding spontaneous CAS episodes, the high-frequency (HF) component decreases while the low-frequency/high-frequency (LF/HF) ratio increases, a pattern consistent with transient sympathetic surges accompanied by vagal withdrawal (28). Microneurography further supports the presence of heightened sympathetic activity in patients with vasospastic angina, suggesting that sympathetic predominance may represent a chronic predisposing state rather than a purely episodic phenomenon (29).
Interventional pharmacological data provide clinical support for ROCK involvement in provoked coronary hyperreactivity. In small clinical studies, the ROCK inhibitor fasudil attenuated acetylcholine-induced coronary constriction and was accompanied by improvement in ischemic findings, consistent with ROCK acting as a downstream mediator of hypercontractile responses in susceptible vessels (30). Taken together, available data suggest that autonomic perturbations, endothelial dysfunction, and VSMCs hypercontractility interact to lower the threshold for CAS initiation and recurrence.
Amplification via feedback: spasm–induced ischemia as a driver of endothelial injury and dysfunctional vicious cycle
4.4
Coronary spasm-induced ischemia may further aggravate endothelial dysfunction and sustain a vicious cycle of recurrent spasm. Preclinical studies and clinical observational data support several interrelated pathways:
- (a)Excessive ROS generation. Ischemia–reperfusion injury increases ROS levels and promotes endothelial damage, further depleting NO and worsening endothelial dysfunction (31).
- (b)Inflammatory cytokine activation. Ischemic insult can activate pro-inflammatory mediators such as TNF-α and IL-6, which may impair endothelial function and augment VSMCs contractility (32).
- (c)Suppression of endothelial repair capacity. Ischemia may reduce endothelial progenitor cell (EPC) mobilization and accelerate endothelial apoptosis, impairing repair capacity (33). Together, these processes can lower the threshold for recurrent coronary spasm by reinforcing endothelial injury, oxidative stress, and inflammation.
Genetic susceptibility: core molecular loci and pathophysiological mechanisms
4.5
Genetic factors contribute to interindividual variability in CAS susceptibility, with reported loci clustering in pathways that regulate NO bioavailability, oxidative stress handling, and vasoconstrictor signaling (genome-wide association studies and candidate-gene analyses). The ALDH2*2 (rs671) missense variant encodes a low-activity aldehyde dehydrogenase, which can increase reactive aldehyde burden and oxidative stress, thereby favoring NO inactivation and vasomotor hyperreactivity (34). This allele is common in East Asian populations but rare in most European populations, a distribution that may partially contribute to ethnic differences in CAS susceptibility (34). Observational studies further suggest gene–environment interplay, with smoking and alcohol exposure amplifying aldehyde-related oxidative stress and associating with higher spasm propensity among carriers (35).
Beyond ALDH2, the eNOS Glu298Asp (rs1799983) polymorphism has been linked to reduced NO signaling and higher vasospasm risk in case–control and cohort studies (36, 37). In East Asian cohorts, RNF213 p.R4810K has been associated with endothelial and smooth muscle stress phenotypes and enrichment among CAS patients, with observational data linking it to adverse ischemic outcomes (38). In some non-Asian populations, EDN1 Lys198Asn (rs5370) has been associated with diffuse epicardial spasm, potentially reflecting enhanced endothelin-1 signaling (39).
Overall, these findings support a genetic contribution to CAS susceptibility, likely through impaired aldehyde detoxification, oxidative stress, disrupted NO signaling, and augmented vasoconstrictor responses.
Central signaling hub: the multilayered regulatory network of ROCK
4.6
As summarized in Figure 2, ROCK can function as a convergent downstream pathway through which diverse upstream triggers translate into enhanced coronary contractility. Experimental and translational studies implicate RhoA/ROCK activation in settings including nicotine exposure, oxidized LDL/lectin-like oxidized LDL receptor-1 (LOX-1) signaling, sympathetic GPCR stimulation, and PVAT-derived adipokines such as chemerin acting via chemerin receptor (CMKLR1) on VSMCs. Once activated, ROCK promotes spasm mainly by increasing Ca²^+^ sensitivity via MLCP inhibition, by impairing endothelial NO signaling through eNOS suppression, and by facilitating a pro-inflammatory milieu that further augments contractile responsiveness. These observations support ROCK as a mechanistically relevant effector and a promising therapeutic target under active investigation in CAS.
Multidimensional regulatory network of the ROCK signaling pathway in CAS. This schematic depicts the major upstream activators and downstream effectors of the RhoA/ROCK signaling pathway that mediate vascular hypercontractility in CAS. Pathological stimuli, including nicotine, oxidized low-density lipoprotein (ox-LDL), sympathetic neurotransmitters, and chemerin, activate the RhoA/ROCK axis through membrane receptors such as the α-adrenergic receptor (α-AR), lectin-like oxidized LDL receptor-1 (LOX-1), and CMKLR1. These signals converge to enhance RhoA-GTP binding and ROCK activation. Activated ROCK promotes coronary vasospasm by inhibiting MLCP activity, resulting in sustained myosin light chain (MLC) phosphorylation and increased vascular smooth muscle contraction. In parallel, ROCK suppresses eNOS activity, reduces NO production, and induces pro-inflammatory cytokines and adhesion molecules that perpetuate endothelial dysfunction. This network illustrates how environmental, metabolic, and neurohumoral stimuli converge on ROCK signaling to drive CAS pathophysiology and identifies potential pharmacological checkpoints for intervention. MLCP, myosin light chain phosphatase; LOX-1, lectin-like oxidized LDL receptor-1; CMKLR1, chemerin receptor; α-AR, α-adrenergic receptor; eNOS, endothelial nitric oxide synthase; MLC, myosin light chain; ROCK, Rho-associated coiled-coil containing protein kinase.
Risk factor landscape
5
To facilitate critical appraisal, Table 1 summarizes major CAS risk factors, proposed mechanisms, and a structured indication of evidentiary strength (observational, mechanistic, or interventional where available). Ethnic distributions and risk associations of CAS-related genetic polymorphisms are summarized in Table 2.
Smoking: the most well–defined modifiable risk factor for CAS
5.1
Smoking is the most consistently reported modifiable risk factor for CAS. Provocation-tested cohorts, particularly from East Asia, show higher smoking prevalence among CAS patients with dose–response relationships between cumulative exposure and spasm susceptibility (40–42), and smoking is widely regarded as the best-established environmental risk factor (43). Mechanistically, nicotine and combustion-derived oxidants reduce NO bioavailability via oxidative stress, eNOS uncoupling/ADMA-related pathways, and ROCK activation (44–46), and acute exposure may trigger spasm (43). Although CAS-specific cessation trials are lacking, cessation improves vascular function and reduces long-term cardiovascular risk in broader populations (47). it remains a central preventive measure in suspected or confirmed CAS.
Dyslipidemia: the role of oxidative modification in pathophysiology
5.2
Conventional dyslipidemia shows heterogeneous and generally modest associations with CAS (40). In contrast, oxidative lipid pathways—ox-LDL, Lp(a), and dysfunctional HDL—are more consistently linked to endothelial dysfunction and VSMCs hyperreactivity (48, 49). LDL oxidation can promote endothelial injury via NADPH oxidase activation and eNOS uncoupling (50), while ox-LDL can amplify contractile signaling through RhoA-related pathways (51). Statins may improve endothelial function and outcomes in selected VSA/CAS populations (52), but evidence for reducing spasm frequency and the role of Lp(a)-targeted strategies remain limited.
Inflammatory burden and hs–CRP: a reproducible association best interpreted as a risk marker
5.3
Multiple observational studies report associations between elevated hs–CRP and ACh-induced spasm (3, 53, 54), although effect sizes vary and interactions with sex/metabolic status have been described (55). Mechanistic data support plausibility through impaired NO signaling and ROCK-related facilitation of hyperreactivity (56), and PVAT inflammation may further modulate local vasomotor balance (57). Overall, hs–CRP is best interpreted as a risk marker rather than a uniform causal driver.
Renal-linked biomarkers and systemic vulnerability
5.4
In addition to inflammatory markers, renal-linked biomarkers may capture a broader “systemic vulnerability” milieu in which coronary vasomotor dysfunction becomes more likely. Cystatin C has been reported to associate with acetylcholine-provoked CAS in observational cohorts, suggesting that subtle impairment in renal filtration—or the inflammatory, oxidative, and endothelial perturbations that often accompany it—may track with heightened spasm susceptibility (58, 59). Importantly, these data are best interpreted as associative rather than causal: cystatin C integrates multiple upstream processes (renal function, vascular inflammation, and metabolic stress) and therefore remains vulnerable to residual confounding. From a precision-management perspective, its value is less as a standalone discriminator and more as a complementary feature within multimodal risk profiling, helping to contextualize vasomotor testing results and to identify patients whose risk likely reflects a diffuse, multi-organ substrate rather than an isolated coronary phenotype (58, 59).
Drug/toxin exposure: key clinical medication warnings in CAS
5.5
Sympathomimetic exposures (e.g., cocaine) can provoke coronary vasoconstriction via adrenergic stimulation and endothelial dysfunction, contributing to ET-1/NO imbalance and ischemic events (60, 61). Non-selective β-blockers may aggravate vasoconstriction through unopposed α-adrenergic signaling (62), whereas β_1_-selective blockers appear less likely to worsen vasospasm (63). Immunosuppressive and chemotherapeutic agents (e.g., tacrolimus, 5-fluorouracil) have been linked to reversible vasospasm during exposure (64, 65). Medication review and avoidance of recognized triggers remain pragmatic measures.
Myocardial bridging: an anatomical substrate associated with spasm susceptibility
5.6
Myocardial bridging (MB) is associated with increased spasm susceptibility and contributes to a subset of MINOCA presentations (66, 67). Disturbed shear stress within bridged segments may impair endothelial function (including reduced eNOS expression) and promote local hyperreactivity (68). Provocation testing and intravascular imaging suggest that spasm may localize to or adjacent to bridged segments (67). In MB with unexplained angina or MINOCA, concomitant CAS should be considered, and coronary function testing may be useful where available.
Alcohol intake: a controversial factor with genetic and dose-specific modulation
5.7
The relationship between alcohol intake and CAS is heterogeneous. Early reports described alcohol-induced spasm and experimental work suggested vasoconstrictive thresholds (69, 70). Subsequent observations indicate delayed spasm after intake, implicating indirect mechanisms such as endothelial dysfunction and autonomic dysregulation (71, 72). Proposed pathways include altered prostaglandin balance, impaired cGMP-mediated vasodilation, and magnesium depletion affecting eNOS activity (73–76), although these remain hypothesis-generating. ALDH2*2 carriers may be more susceptible to alcohol-related CAS (77). Individualized counseling is appropriate, particularly in patients reporting alcohol-related episodes (78–81).
Age: endothelial senescence and cumulative vascular vulnerability
5.8
Advancing age is associated with higher CAS detection in provocation-tested cohorts (82). Endothelial senescence, cumulative oxidative stress, and low-grade inflammation may lower the threshold for vasomotor dysregulation (83–85), and remodeling/subclinical atherosclerosis may further reduce endothelial resilience. These data support considering CAS in older patients with unexplained ischemia, while optimal screening strategies remain uncertain (82, 86).
Glucose metabolism disorders: metabolic dysregulation rather than overt diabetes as a vasomotor modifier
5.9
Associations between diabetes and CAS are inconsistent across provocation cohorts (87–89). In contrast, insulin resistance and compensatory hyperinsulinemia may impair NO signaling and microvascular vasodilation, contributing to vasomotor dysregulation even without overt diabetes (88, 90). Follow-up studies also suggest higher incident diabetes in CAS populations (91). Clinically, assessing metabolic status and monitoring for incident diabetes remain appropriate (92, 93).
Hypertension: paradoxical association with CAS susceptibility
5.10
Although hypertension contributes to endothelial dysfunction, its relationship with CAS differs from ASCVD. Several provocation-based studies report neutral or inverse associations between hypertension and ACh-induced spasm (3, 94). Proposed mechanisms include remodeling and altered calcium-handling/ROCK signaling that may reduce acute pharmacologic responsiveness in certain contexts (95–98). Hypertension should be treated for overall cardiovascular risk reduction, while its role as a CAS-specific risk factor should be interpreted cautiously.
Hyperuricemia: a marker of spasm severity rather than incidence
5.11
Studies on serum uric acid show mixed findings, with inconsistent associations with CAS occurrence but reported links with multivessel spasm in some cohorts (99, 100). Mechanistic studies suggest effects on eNOS, oxidative stress, and endothelin/ROCK pathways (101–103), but CAS-specific interventional confirmation is lacking. Hyperuricemia is therefore best viewed as a potential marker of severity rather than a therapeutic target.
Gender differences and phenotypic heterogeneity
5.12
Men more frequently exhibit epicardial spasm, whereas women more often demonstrate microvascular spasm in multicenter observational studies and meta-analyses (104, 105). Estrogen may enhance NO signaling and suppress vasoconstrictor pathways, and post-menopausal changes may contribute to instability (106, 107), although hormone-based interventions remain unproven. Sex-specific interactions with smoking, aging, inflammatory, and metabolic factors have also been reported (54, 108), supporting sex-informed evaluation rather than sex-specific therapy.
Physiological and psychological stress: autonomic dysregulation as a trigger
5.13
Autonomic imbalance is frequently observed in vasospastic angina. Heart-rate variability and Holter studies show short-term sympathetic–parasympathetic shifts preceding spasm episodes (109–111). These data are observational and do not establish causality. Where CAS coexists with structural coronary disease, autonomic dysfunction has been associated with worse prognosis (112). Psychological stress may also contribute: mental stress testing can provoke ischemic changes consistent with vasospasm (113), and cohort studies associate anxiety/depression with higher CAS prevalence (114–116), although CAS-specific randomized trials of stress-reduction interventions are lacking.
Environmental factors: external triggers of vasomotor instability
5.14
Environmental exposures may act as external triggers that destabilize vasomotor tone in predisposed individuals. Higher long-term PM₂.₅/PM₁₀ exposure has been associated (in multivariable models) with ACh-provocation positivity, with PM₂.₅ showing a stronger relationship with epicardial spasm and both pollutants linked to MINOCA presentations (117). Mechanistic data support plausibility through systemic inflammation, oxidative stress, and downstream ROCK activation, promoting vascular hyperreactivity (118). Cold exposure and hyperventilation are recognized triggers; provocation studies report high sensitivity of combined cold-pressor plus hyperventilation protocols for eliciting variant angina–type responses (119). Overall, the evidence supports an “environmental trigger” model, while causal inference remains limited by exposure assessment and residual confounding.
Ethnicity and genetic susceptibility
5.15
Ethnic differences in CAS prevalence are consistently reported, with higher detection rates in East Asian populations than in Western cohorts (104, 120). Genetic studies implicate variants in ALDH2, EDN1, and other vasomotor-related genes as susceptibility loci (39, 77, 121), supporting gene–environment interaction rather than single-gene causation. At present, genetic testing has no established role in routine CAS management.
Comorbid diseases: systemic vasomotor dysregulation
5.16
Several comorbid conditions have been associated with CAS, plausibly reflecting systemic vasomotor vulnerability.
Obstructive sleep apnea (OSA): Observational studies report higher acetylcholine-provocation positivity in OSA, and CPAP improves endothelial function (122–124). However, CAS-specific interventional trials are unavailable.
Thyrotoxicosis: Case reports and small series describe reversible coronary spasm resolving after restoration of euthyroidism (125, 126). Evidence remains largely observational.
Migraine and Raynaud's Phenomenon: Both represent vasomotor hyperreactivity phenotypes, and observational studies report higher prevalence among CAS patients (127–129). Shared genetic predisposition has been suggested.
Collectively, these comorbidities are best viewed as markers of systemic vasomotor susceptibility rather than direct causal factors.
Summary and future directions
6
CAS is a multifactorial vasomotor disorder in which endothelial dysfunction and VSMCs hyperreactivity interact with inflammation–oxidative pathways, autonomic imbalance, and genetic susceptibility. While parts of the risk profile overlap with ASCVD (e.g., smoking and dyslipidemia), CAS also has distinct modifiers, including myocardial bridging, PVAT-related dysfunction, and population-specific genetic variants that may shape susceptibility and clinical expression.
Key strategies to improve prognosis include
7
Risk-factor modification
7.1
Smoking cessation remains the most actionable preventive intervention, alongside mitigation of inflammatory burden, avoidance of relevant triggers (including medications and environmental exposures), and optimization of comorbidities such as OSA.
Phenotype-informed therapy and evidence gaps
7.2
Calcium channel blockers remain first-line therapy. For refractory cases, small interventional studies suggest that ROCK inhibition (e.g., fasudil) can attenuate ACh-induced constriction and ischemic changes, including in microvascular spasm phenotypes (30, 130). However, robust CAS-specific outcome trials—particularly phenotype-stratified comparisons between epicardial and microvascular spasm—remain limited. Therefore, phenotype-guided pharmacotherapy should be presented as a rational but still unvalidated strategy that requires prospective confirmation.
Priorities for future research
7.3
(1)Define how major modifiers (e.g., ALDH2*2, tobacco exposure, PM₂.₅) interact within the “amplification loop,” and identify tractable regulatory nodes.(2)Determine whether multimodal tools (e.g., leukocyte ROCK activity, PVAT attenuation, genetic risk scores, renal-linked biomarkers) improve risk stratification and treatment selection beyond clinical phenotyping alone; cystatin C associations require external validation (58, 59).(3)Clarify endotype overlap and diagnostic uncertainty across epicardial and microvascular spasm, and establish standardized, reproducible testing thresholds to reduce misclassification and inappropriate therapeutic generalization.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Barsukov AV Borisova EV Vasilyeva IA Glukhovskoy DV Kuznetsov MV Saraev GB. Vasosspastic angina: current concepts about pathogenesis, diagnosis, treatment (literature review). Adv Gerontol. (2025) 38(1):36–45.40570213 · pubmed ↗
- 2Saito S Yamagishi M Takayama T Chiku M Koyama J Ito K Plaque morphology at coronary sites with focal spasm in variant angina: study using intravascular ultrasound. Circ J. (2003) 67(12):1041–5. 10.1253/circj.67.104114639021 · doi ↗ · pubmed ↗
- 3Morita S Mizuno Y Harada E Nakagawa H Morikawa Y Saito Y Differences and interactions between risk factors for coronary spasm and atherosclerosis–smoking, aging, inflammation, and blood pressure. Intern Med. (2014) 53(23):2663–70. 10.2169/internalmedicine.53.270525447646 · doi ↗ · pubmed ↗
- 4Jenkins K Pompei G Ganzorig N Brown S Beltrame J Kunadian V. Vasospastic angina: a review on diagnostic approach and management. Ther Adv Cardiovasc Dis. (2024) 18:17539447241230400. 10.1177/1753944724123040038343041 PMC 10860484 · doi ↗ · pubmed ↗
- 5Montone RA Rinaldi R Del Buono MG Gurgoglione F La Vecchia G Russo M Safety and prognostic relevance of acetylcholine testing in patients with stable myocardial ischaemia or myocardial infarction and non-obstructive coronary arteries. Euro Intervention. (2022) 18(8):e 666–e 76. 10.4244/EIJ-D-21-0097135377315 PMC 10241282 · doi ↗ · pubmed ↗
- 6Montone RA Cosentino N Gorla R Biscaglia S La Vecchia G Rinaldi R Stratified treatment of myocardial infarction with non-obstructive coronary arteries: the PROMISE trial. Eur Heart J. (2025):ehaf 917. 10.1093/eurheartj/ehaf 91741150941 PMC 13008472 · doi ↗ · pubmed ↗
- 7Ong P Camici PG Beltrame JF Crea F Shimokawa H Sechtem U International standardization of diagnostic criteria for microvascular angina. Int J Cardiol. (2018) 250:16–20. 10.1016/j.ijcard.2017.08.06829031990 · doi ↗ · pubmed ↗
- 8Ong P Athanasiadis A Borgulya G Vokshi I Bastiaenen R Kubik S Clinical usefulness, angiographic characteristics, and safety evaluation of intracoronary acetylcholine provocation testing among 921 consecutive white patients with unobstructed coronary arteries. Circulation. (2014) 129(17):1723–30. 10.1161/CIRCULATIONAHA.113.00409624573349 · doi ↗ · pubmed ↗