The role of histone modifications in transcription regulation upon DNA damage
Angelina Job Kolady, Siyao Wang

TL;DR
This review explains how changes to histone proteins help control gene activity during DNA damage, which is important for preventing diseases like cancer.
Contribution
The paper provides a comprehensive overview of how specific histone modifications regulate transcription during DNA damage response.
Findings
Histone modifications like phosphorylation and acetylation control DNA accessibility for repair.
Dysregulation of these modifications is linked to cancer and neurodegenerative diseases.
Understanding these mechanisms can guide the development of new therapeutic strategies.
Abstract
Cells are constantly exposed to various sources of DNA damage, including radiation, chemicals, replicative stress and oxidative stress, that threaten genome stability. To ensure faithful DNA repair, transcription regulation needs to be tightly controlled. This regulation involves transcriptional suppression, selective activation of DNA repair‐related genes and transcriptional recovery post‐repair. Failure to properly modulate transcription during DNA damage can result in collisions between transcriptional and repair machineries, misregulation of repair genes and delayed recovery, ultimately compromising genomic integrity. Chromatin modifications play a central role in this process. These modifications include phosphorylation, methylation, acetylation and ubiquitination, which orchestrate DNA accessibility for repair machinery and fine‐tune transcriptional responses. Absence of these…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
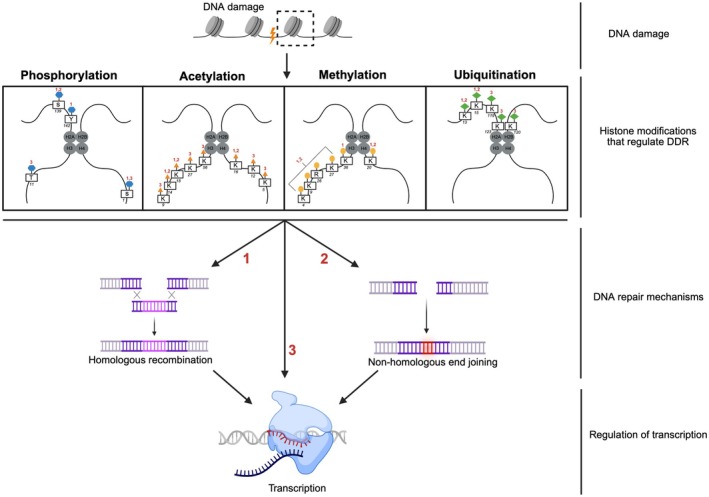 Fig. 1
Fig. 1| Histone | Amino acid residue | Modification | Function after DNA damage | Function in transcription upon DNA damage |
|---|---|---|---|---|
| H2A | Tyrosine 142 | Phosphorylation | Recruits RAD51 for transcription‐coupled HR | Transcription initiation |
| H2AX | Serine 139 | Phosphorylation | Forms MRN complex and initiates DNA repair | Transcription initiation and elongation |
| H2A | Lysine 123 | Ubiquitination | Phosphorylates RAD9 | Regulates transcription elongation |
| H2A | Lysine 119 | Ubiquitination | Releases RNA pol II | Transcription pause‐release |
| H2A | Lysine 15 | Ubiquitination | Recruits 53BP1 | Transcription stalling and represses initiation and elongation |
| H2A | Lysine 13 | Ubiquitination | Recruits 53BP1 | Transcription stalling and represses initiation and elongation |
| H2B | Lysine 120 | Ubiquitination | Recruits FACT complex | Transcription elongation |
| H3 | Lysine 122 | Succinylation | Activates transcription and deposits at TSS | Transcription initiation |
| H3 | Lysine 56 | Acetylation | Can promote rapid nucleosome displacement | Transcription initiation |
| H3 | Lysine 36 | Methylation | Recruits YKU70 and maintains HR | Transcription elongation and recovery |
| H3 | Lysine 27 | Acetylation | Regulates levels of sirtuins and maintains transcription levels | Transcription initiation, elongation and recovery |
| H3 | Lysine 18 | Acetylation | Downregulates 53BP1 | Transcription initiation and recovery |
| H3 | Lysine 14 | Acetylation | Retention of RSC and regulates DNA repair | Transcription initiation and elongation |
| H3 | Threonine 11 | Phosphorylation | Regulate gene expression | Transcription initiation |
| H3 | Serine 10 | Phosphorylation | Activates transcription | Transcription initiation and recovery of elongation |
| H3 | Lysine 9 | Acetylation | Can promote NHEJ by upregulating expression of DNA repair factors | Transcription initiation |
| H3 | Lysine 9 | Methylation | Regulates expression of DNA repair factors | Transcriptional silencing or repression |
| H3 | Lysine 4 | Methylation | Recruits yKu proteins and promotes NHEJ | Transcription initiation and elongation and recovery |
| H4 | Lysine 16 | Acetylation | NHEJ and HR repair | Transcription initiation and elongation |
| H4 | Lysine 12 | Acetylation | Recruit BRD2 to DSBs for repair | Transcription elongation |
| H4 | Lysine 5 | Acetylation | Reassembly of chromatin after DNA repair | Transcription regulation |
| H4 | Serine 1 | Phosphorylation | Re‐ligation of strands in NHEJ pathway | Transcription elongation |
- —Federation of European Biochemical Societies10.13039/100012623
- —Deutsche Forschungsgemeinschaft10.13039/501100001659
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA Repair Mechanisms · Epigenetics and DNA Methylation · Telomeres, Telomerase, and Senescence
Abbreviations
ART, ADP‐ ribosyltransferase
ATM, ataxia‐telangiectasia‐mutated
ATP, adenosine tri phosphate
ATR, ataxia – telangiectasia and Rad3‐related
BER, base excision repair
BRCT, BRCA 1 terminus
BRD4, bromodomain‐containing protein 4
CAT, catalytic domain
CDK, cyclin‐dependent kinase
CS, Cockayne syndrome
CTD, C‐ terminal domain
DDR, DNA damage response
DNA, deoxyribonucleic acid
DOT1L, disruptor of telomeric silencing 1‐like
DPC, DNA–protein crosslink
DSB, double‐stranded break
DSIF, DRB sensitivity‐inducing factor
ERK, extracellular signal‐regulated kinase
EZH2, enhancer of zeste homologue 2
FACT, facilitating chromatin
HAT, histone acetyl transferase
HD, helical domain
HDAC, histone deacetylase
KAP 1, Krüppel‐associated box domain‐associated protein‐1
MDC1, mediator of DNA damage checkpoint protein 1
MRE11, meiotic recombination 1
NBS1, Nijimegen breakage syndrome 1
NELF, negative elongation factor
NER, nucleotide excision repair
NHEJ, non‐homologous end joining
NuRD, nucleosome remodelling and deacetylase
PARP, poly ADP Ribose polymerase
PcG, polycomb group
PD, Parkinson's disease
PIC, pre‐initiation complex
PKA, protein kinase A
PRC, polycomb repressive complex
P‐TEFb, positive transcription elongation factorb
PTM, post‐translational modification
RNA Pol II, RNA polymerase II
SAM, S‐adenosyl‐methionine
SET 1D, SET domain containing 1A
SSB, single‐stranded break
TBL, transcription‐blocking lesion
TBP, TATA–box binding protein
TF, transcription factor
TTD, trichothiodystrophy
VRK1, Vaccinia‐related kinase 1
WIP, wild‐type phosphatase
XPC, Xeroderma pigmentosum complementation group C
XRCC1, X ray repair cross‐complementing protein 1
DNA, the basic unit of genetic information, is approximately two metres long and is tightly packed around positively charged histone proteins. Histones bind electrostatically to the negatively charged phosphate groups of DNA, forming nucleosomes, the basic unit of chromatin. Each nucleosome consists of an octameric core of histones (H2A, H2B, H3 and H4), while linker histone H1 (in eukaryotes) and H5 (a variant of H1, found in avian and reptilian species) stabilize higher‐order chromatin conformation [1]. This compact structural state of chromatin restricts access to DNA and thus modulates various biological processes, including transcription, replication and DNA repair [2, 3].
Chromatin accessibility is dynamically regulated by post‐translational modifications (PTMs) on histones, which either serve as docking platforms for chromatin remodellers and histone chaperones or alter their electrostatic properties [4, 5]. These modifications, which include PARylation, phosphorylation, acetylation, methylation, SUMOylation, crotonylation, succinylation, isomerization and citrullination, play a pivotal role in DNA‐related functions. One of their major roles is regulating transcription in response to DNA damage, ensuring transcription recovery and genome stability.
Transcription regulation in eukaryotes
In eukaryotes, transcription initiation begins with TFIID binding to the TATA sequence of the DNA via the TBP (TATA‐binding protein) subunit, followed by the sequential recruitment of general transcription factors (TFIIA, TFIIB, TFIIE, TFIIF and TFIIH) and RNA polymerase (Pol II) [6], finally forming the pre‐initiation complex (PIC) to initiate the synthesis of RNA using DNA as a template [7]. Following initiation, Spt5/4 (DSIF, DRB sensitivity‐inducing factor) and negative elongation‐inducing factor (NELF) transiently pause RNA Pol II. Transcription elongation is then triggered by the phosphorylation of RNA Pol II C‐terminal domain (CTD) at serine 2 by the catalytic cyclin‐dependent kinase 9 (CDK9), a subunit of the positive transcription elongation factor (P‐TEFb) [8, 9].
The tight association of histone tails with DNA presents a physical barrier to the progression of RNA Pol II. However, histone PTMs and chromatin remodellers, such as facilitates chromatin transcription (FACT) complex, can loosen the contact between DNA and histone, therefore facilitating RNA Pol II passage and later reassembly of the nucleosome [10, 11, 12].
Transcription regulation in the DNA damage response (DDR)
Transcription inhibition has been recognized as an indicator of DNA damage because DNA lesions physically obstruct ongoing transcription. Conversely, the recovery of transcription has been considered a surrogate marker of DNA repair. However, the relationship between DNA damage, DNA repair and transcription is much more complex due to the various direct and indirect effects of DNA damage on transcription.
The transcriptional outcomes following DNA damage depend on the type and location of the DNA lesion. Non‐bulky lesions such as the oxidative base lesion 8‐oxoguanine (8‐oxoG), DNA nicks, or single‐stranded gaps can be bypassed by the RNA Pol II machinery, leading to transcriptional mutagenesis and affecting cellular function. When such mutations occur within a gene regulatory sequence, they could prevent the binding of transcription factors, resulting in the misregulation of the target genes [13]. In contrast, helix‐distorting bulky DNA lesions or cyclobutene pyrimidine dimers (CPDs) occurring in the transcribed region of genes, caused by UV radiation or cross‐linking agents, can stall RNA Pol II progression, triggering a cascade of nucleotide excision repair (NER) and transcriptional recovery events. Defects in factors responsible for the removal of transcription‐blocking lesions (TBLs) can lead to various disorders, such as Cockayne syndrome (CS), trichothiodystrophy (TTD), neurodegenerative diseases, infectious diseases and inflammation, highlighting the importance of understanding DNA damage‐induced transcription regulation [14, 15, 16, 17, 18]. The Cockayne syndrome B (CSB) protein, which is a DNA‐dependent ATPase, promotes RNA Pol II progression under normal conditions but fails to resolve TBLs, leading to transcriptional suppression [19]. Subsequent recruitment of the CSA‐DDB1‐CRL4 ubiquitin ligase complex facilitates RNA Pol II ubiquitylation on K1268 of the RPB1 subunit, serving as a marker for repair [20, 21]. The DNA repair factor ultraviolet‐stimulated scaffold protein A (UVSSA) then mediates RNA Pol II backtracking, enabling the exposure of DNA lesions and recruits the NER machinery, including TFIIH [22, 23]. TFIIH not only facilitates DNA repair but also plays a prominent role in transcriptional recovery through CDK7‐mediated phosphorylation on serine 5 of RNA Pol II [24]. Additionally, the FACT complex assembles the nucleosome for the RNA Pol II to move forward, thus aiding in transcription elongation recovery [25, 26].
Another major cause of transcription stalling is DNA–protein crosslinks (DPCs). These covalent adducts act as a barrier to the progression of RNA polymerase II transcription and stall DNA replication. Stalling of RNA Pol II after DPC formation initiates transcription‐coupled (TC) DPC repair by recruiting CSB and CSA proteins [27, 28, 29, 30]. To ensure proper replication after DPC in mammals, SPRTN, a replication‐specific protease, digests the proteinaceous part of the DPC, protecting from the replication fork collapse [31]. DNA damage can also occur during the process of transcription itself. Nascent RNA binds to the template DNA strand forming a short DNA–RNA hybrid or a three‐stranded structure, known as an R‐loop. These structures accumulate during transcription initiation and termination, and block the progression of RNA Pol II, causing DNA damage and threatening genome stability [30]. The TC‐NER mechanism, and other enzymes such as RNase H, helicases and topoisomerases are involved in resolving these R‐loop structures and maintaining genome integrity [32].
DNA damage can regulate transcription both in cis and in trans. Many DDR genes, including the tumour suppressor protein p53, are activated in response to the blockage of transcription. Following its activation, p53 accumulates in the nucleus and functions as a transcription factor to induce the transcription of downstream genes. Depending on the cell type, damage type and severity, p53 can enhance cell survival by upregulating DNA repair genes and cell cycle inhibitors or induce cell death by activating apoptotic genes. In both scenarios, the mutagenesis and cytotoxicity caused by DNA damage can be mitigated [33].
Due to the role of histone modifications in regulating gene expression, DNA repair and chromatin architecture, their dysregulation can contribute to various disorders such as cancer, neurodegenerative diseases, ageing and other metabolic disorders [34, 35, 36]. Several examples, such as uncontrolled H3 and H4 acetylation, can influence transcription silencing, contributing to Huntington's disease. Loss of H4K16ac contributes to defective DNA repair, resulting in Werner's syndrome and progeria. Impaired γ‐H2AX at the site of DNA damage due to mutated ATM can cause ataxia‐telangiectasia. Abnormal histone acetylation levels are observed in peripheral blood mononuclear cells of Type 2 diabetes patients, suggesting this modification's involvement in the pathogenesis of metabolic diseases. These studies suggest the role of histone modifications in DNA damage‐related pathogenesis.
This review explores the role of histone modifications in regulating transcription during the DDR Fig. 1, with a focus on their mechanistic contributions to damage recognition, repair and transcriptional recovery. We also discuss the implications of dysregulated histone PTMs in ageing‐related diseases and highlight emerging modifications in DDR that warrant further investigation.
Schematic representation of various types of histone modifications in transcription regulation during the DDR. Created in BioRender. Wang, S. (2026) https://BioRender.com/4gf9os7.
Histone PARylation
Histone poly(ADP‐ribosyl)ation or histone PARylation is a post‐translational modification where ADP ribose polymers are attached to histone proteins. This modification is primarily catalysed by Poly ADP Ribose polymerases (PARP). The catalytic domain (CAT) of PARP proteins binds nicotinamide adenine dinucleotide (NAD+) catalysing the PARylation of target proteins, including histones [37]. The PARP family of proteins consists of 17 members, out of which PARP1 and PARP2 are the most abundant forms involved in DNA damage, replication, transcription and apoptosis [38]. PARP‐1 contains 6 domains including two homologous zinc finger domains (Zn1 and Zn2) followed by a Zn3 and a WGR (Trp‐Gly‐Arg) domain [38, 39]. Between the Zn3 and the WGR domains, there is the auto‐modification domain (AD) which contains the BRCA1 C terminus (BRCT) fold that recognizes intact DNA and mediates the interaction with DNA repair proteins [40]. Functionally, it facilitates DNA repair by stimulating base excision repair (BER), non‐homologous end joining (NHEJ) and homologous recombination (HR) [41].
Histone PARylation in the DDR
During DNA damage, PARP's Zn1 and Zn2 domains recognize the damage site by forming either a ‘base stacking loop’ with the exposed nucleotides or a ‘backbone grip’ with the DNA phosphate backbone [41]. This interaction causes a conformational change in PARP's CAT domain, making NAD+ accessible and forming negatively charged PAR chains at DNA damage sites [33]. PAR chains can open the chromatin and enable DNA repair, by recruiting proteins such as X‐ray repair cross‐complementing protein 1 (XRCC1) [42], xeroderma pigmentosum C (XPC) [43] and meiotic recombination 11 (MRE11) or Nijimegan breakage syndrome 1 (NBS1) [44] in single‐stranded breaks (SSBs), UV‐induced photolesions and double‐strand breaks (DSBs), respectively. In the presence of DSBs, PARP1 plays a crucial role in regulating DNA repair pathway choice. At the DSB site, PARP1 competes with the Ku70/80 complex during the S/G2 phases, promoting alternative NHEJ, or interacts with DNA‐PKcs, resulting in classical NHEJ [45]. Reports also show that MRE11 interacts non‐covalently with PAR chains, which is essential for HR [45].
Excessive PARP activity leads to NAD + depletion due to its over consumption, leading to neurological and mitochondrial dysfunction, as well as ageing‐associated diseases [46, 47, 48]. Thus, it is essential to suppress PARP activity after DNA lesion recognition. Recent reports suggest that the functions of PARP1 and XRCC1 are reciprocal upon genotoxic stress. To prevent overactivation of PARP activity and restore chromatin stability after DNA repair, the recruitment of XRCC1 dissociates PARP from the DNA [49]. Lee et al. [50] show that the Ewing sarcoma protein (EWS) is required for the proper dissociation of PARylated PARP1 from DNA damage sites after the recruitment of the DNA repair proteins. Thus, auto‐PARylation and PARylation of other proteins is a highly regulated process involved in the maintenance of genomic stability.
PARylation in transcriptional regulation during the DDR
The PARP protein family plays a vital role in regulating transcription and maintaining genome integrity, by chromatin remodelling [51, 52], interacting with transcription factors, displacing the RNA polymerase machinery during damage and through crosstalk with other post‐translational modifications [53].
Immediately after DNA damage, it is important to pause ongoing transcription to prevent collisions between the transcriptional and DNA repair machineries [54]. PARP1 auto‐PARylation produces PAR structures that recruit PcG and NuRD complexes to the sites of DNA damage [55, 56]. These transcriptional repressor complexes evict RNA Pol II from the chromatin to prevent inefficient transcription and the release of premature transcripts [55, 57]. Another report suggests that PARP1 binds to P‐TEFb upon DNA damage, preventing cyclin—T1 from undergoing liquid–liquid phase separation thereby stopping CDK9 from phosphorylating RNA Pol II serine 2 and resulting in transcription elongation inhibition [57]. Therefore, PARP1 acts as a quality control factor to prevent the release of immature transcripts.
On the contrary, PARP1 regulates remodelling of the chromatin and recruitment of DNA repair factors at the damaged sites to facilitate transcription in cis. Immediately upon DNA damage, PARP1 is activated and it PARylates the KDM5B demethylase for H3K4me3, excluding KDM5B and opening the chromatin to allow DNA repair [58]. Meanwhile, PARylation of PARP1 can also dissociate H1 from the promoter regions of the genes. Dissociation of H1, a repressor of RNA polymerase II‐mediated transcription, facilitates active chromatin for DNA repair and thereby active transcription [59]. At the time of DNA damage, the KDM4D demethylase is PARylated on its C‐terminal domain, to the site of DNA damage, thereby increasing the demethylation activity of H3K9, facilitating open chromatin and promoting DSB repair [60, 61, 62] in the neurons of the hippocampus and amygdala.
Beyond the role in chromatin remodelling, PARP interacts with various transcription factors to modulate the transcriptional activation of DDR genes. Post DNA damage, the NF‐κB p65‐p50 heterodimer is generally translocated to the nucleus [63, 64]. PARP1‐mediated PARylation at the damage site causes the release of the negatively charged PAR chains that directly bind p65, stabilizing the interaction between NF‐κB and the transcription machinery [18]. This facilitates the pre‐initiation complex formation and transcription activation, thereby enhancing transcription initiation of NF‐κB‐dependent anti‐apoptotic genes [64]. PARP1 also acts as a transcription co‐activator via interacting with or PARylating various transcription factors. For instance, it physically binds the promoter of the transcription factor E2F and upregulates its expression [65]. E2F, in turn acts as a transcription activator after being phosphorylated by ATM or ATK, recruits XPA to the site of damage and facilitates the retention of other repair factors [66, 67]. PARylation generally produces a dual response either by regulating transcription factors or cell cycle checkpoints [68]. AutoPARylated PARP1 binds to p53, promoting covalent PARylation and recruiting p53 to the DNA damage site. This paves the way for the recruitment of 53BP1 and DDB1 to the damage and facilitates HR and NHEJ, highlighting the role of PARylation in activating DNA repair and transcription [69]. PARP1 activation upon DNA damage can activate the PLK3 gene in an ATM‐mediated manner and phosphorylate p53, affecting its transcriptional activity [70], essential for p53‐dependent apoptosis [71].
PARylation engages in crosstalk with other histone PTMs to orchestrate the transcriptional response to DNA damage. γ‐H2AX, known as the primary histone marker of DSBs, recruits PARP1 to the damaged site. This association enhances the catalytic activity of PARP1 and initiates the first step of DNA repair [72]. A recent study shows that histone deacetylase 5 (HDAC5)‐mediated histone deacetylation modulates PARP1's DDR and facilitates the efficient recruitment of DNA repair factors following damage [73].
Overactivation of PARP1 causes various ageing‐related disorders, such as osteosclerosis, neurodegeneration, Parkinson's disease and Alzheimer's disease [74, 75]. To prevent excessive activation of PARP1 after DNA repair is completed, RNF146 uses its WWE domain to recognize and bind PAR chains, which leads to the ubiquitination and subsequent degradation of PARP1 [76]. Inhibition of PARP1 can lead to the accumulation of RNA Pol II at the post‐damage sites, indicating the importance of PARylation for efficient transcription recovery [77].
The role of histone PARylation in DDR is highly exploited in cancer therapeutics. FDA‐approved PARP inhibitors such as Olaparib, Rucaparib, Talazoparib and Niraparib are used to block PARP1 activity, thus accumulating DNA damage and causing cell death in breast cancer type 1 susceptibility protein (BRCA 1)‐deficient cancer cells that have a defective HR [78]. PARP inhibitors are also used in combination with chemotherapy, radiotherapy and immunotherapy to enhance the effectiveness of the treatment and overcome the development of resistance to treatment by the tumour cells. Due to the various functions of PARP1 and its associated proteins, it will be interesting to explore how PARP balances its dual role as a transcription activator and repressor upon DNA damage.
Histone phosphorylation
Histone phosphorylation is a process by which protein kinases, such as PKA, CDK and ATR, add negatively charged phosphate groups from ATP to the hydroxyl group of amino acid residues such as serine, threonine, tyrosine on the N terminus of histone tails [5, 79] thereby promoting transcription activation [80]. Histone phosphorylation can alter chromatin structure, serving as a platform to recruit various DNA repair proteins and transcription factors, ensuring genome stability [79].
Histone phosphorylation in the DDR
Among all the histone PTMs, H2AX phosphorylation (γ‐H2AX) serves as the most established biomarker of DNA damage. H2AX phosphorylation at serine 139 is a rapid response that occurs within seconds following DSB generation [81]. Initial damage recognition involves NBS1 recruitment to the damage site through its interaction with the MRN complex (MRE11‐RAD50‐NBS1) [82]. Subsequently, ATM phosphorylates NBS1 and H2AX at their N terminus, while activating downstream transcription and cell cycle check point regulators. γ‐H2AX stabilizes the MRN complex and recruits repair proteins such as 53BP1 and RAD51 to the damage site, thereby initiating NHEJ and HR repair pathways [83, 84]. Notably, during infrared irradiation, Vaccinia‐related kinase 1 (VRK1), a chromatin kinase in cooperation with ATM, phosphorylates H2AX at serine 139 [85]. γ‐H2AX‐deficient mice exhibit growth retardation, chromosome instability and DNA repair deficiency, underscoring the crucial role of γ‐H2AX in genomic maintenance [86].
After DNA repair completion, it is vital to remove the phosphorylation of H2AX to prevent cellular senescence and ageing [87]. Dephosphorylation of γ‐H2AX by wild‐type p53‐induced phosphatase 1 (WIP1) prevents further recruitment of DNA repair proteins such as 53BP1 to the lesion site, ensuring the completion of DNA repair [88]. Another approach to remove γ‐H2AX from the damage site happens through the recruitment of BRCA1. BRCA1 interacts with H2AX to form a stable biochemical complex. This complex facilitates ubiquitination of H2AX at lysine 119, thus attenuating γ‐H2AX level [89]. It is hypothesized that the interaction between BRCA1 and γ‐H2AX ensures timely suppression of the active repair phase. Studies indicate that H4S1 phosphorylation mediated by casein kinase II occurs within 1 kb of the DSB site, after NuA4 activity, which is involved in recruiting DNA repair proteins during NER. Thus, H4S1 phosphorylation after the action of NuA4 and its correlation with chromatin deacetylation, which blocks further action by NuA4, suggests its role in chromatin restoration after DNA repair [90].
Histone phosphorylation in transcriptional regulation during the DDR
Histone phosphorylation is critical for regulating transcription in response to DNA damage, resulting either in transcription attenuation at the damage sites or chromatin remodelling to activate DDR‐related transcription [91]. In addition, it regulates transcription via phosphorylating other histone modifying enzymes in an ATM/ATR‐dependent manner [92].
As described earlier, immediately after DNA damage, γ‐H2AX is recruited to the DSB site and retained due to the autophosphorylation of ATM kinase at serine 1981. Although ATM autophosphorylation is not required for γ‐H2AX, ATM kinase activity itself is still essential [93]. Following γ‐H2AX, mediator of DNA damage checkpoint 1(MDC1) is recruited and binds to γ‐H2AX via its BRCT domain, thereby stabilizing the damage foci [94]. MDC1 in turn recruits RNF8‐E3 ubiquitin ligase, which mono‐ubiquitinates H2A and H2AX, facilitating RNF168 binding and enhancing ubiquitination [79]. This amplified γ‐H2AX‐mediated ubiquitination signal serves as a platform for transcription repressor complexes to bind and silence transcription at the damage site [95]. Evidence suggests that the absence of H2A ubiquitylation, caused by mutated RNF8, can rescue transcription [95].
The dissociated 53BP1 from the Tudor‐interacting repair regulator (TIRR)/53BP1 complex [96] binds to the ubiquitinated H2AX foci at the damaged site, leading to the RNA Pol II dissociation. Mechanistically, 53BP1 engages in crosstalk with H4K20me2 via its Tudor domain [97], leading to chromatin compaction and preventing the RNA Pol II machinery from moving forward, thereby causing its dissociation from the chromatin [98, 99, 100]. In addition, 53BP1 can further enhance chromatin compaction and RNA Pol II dissociation by stabilizing histone deacetylase 1 (HDAC1) [98]. These pathways play a key role in regulating transcription by compacting chromatin in a timely manner and arresting transcription elongation at damaged sites. After the completion of DNA repair, γ‐H2AX is dephosphorylated by PP2A phosphatases, preventing transcriptional silencing and thus promoting transcriptional recovery [101].
Beyond the γ‐H2AX‐mediated local transcriptional silencing at damaged sites, other histone phosphorylation events can activate the transcription of certain DDR genes. For instance, ERK kinase‐mediated H3S10 phosphorylation induces chromatin decompaction and activates the transcription of DDR genes immediately following genotoxic stress [102]. It was shown that H3S10 phosphorylation negatively correlates with H3K9me2, a mark for transcriptional repression, while co‐occurring with active transcription marks including H3K4me3, H3K9ac and H3K27ac. Thus, H3S10 phosphorylation associates with enhanced transcription elongation [103]. Additionally, H3S28 phosphorylation activates the transcription factors such as C‐FOS and JUN following UV‐induced DNA damage or other stressors [104].
Aberrant histone phosphorylation is associated with genome instability and several disorders [105]. Studies show that elevated levels of γ‐H2AX are associated with neurodegenerative diseases [106]. γ‐H2AX is also a widely used biomarker of DNA damage in cancer diagnosis and treatment. A significant increase in γ‐H2AX expression is found in gastric carcinoma [107], lung cancer [108], melanoma [109] and colon cancer [110]. In colorectal cancer, γ‐H2AX expression highly correlates with the tumour's malignant behaviour and poor survival of the patients [111]. Importantly, γ‐H2AX is not always ‘bad’; in fact, it represents a necessary and beneficial step in the DDR of healthy cells. However, the role of γ‐H2AX is highly context‐dependent. In pre‐cancer cells, if DNA damage persists and γ‐H2AX foci remain unresolved, this persistence can promote carcinogenesis by promoting genomic instability. Once a tumour is established, γ‐H2AX accumulation can act in two opposing ways. On one hand, it can be advantageous, since sustained signalling may cause replication arrest or trigger cell death, thereby suppressing tumour growth. On the other hand, it can also be detrimental, as chronic γ‐H2AX signalling may contribute to further mutations and genomic rearrangements that enhance the metastatic potential of cancer cells. Precancerous lesions have high levels of DSBs due to active oncogenes stalling DNA replication forks [112]. Thus, due to the levels of DSBs, γ‐H2AX is used as a biomarker to detect these lesions and take preventive measures. Moreover, telomere shortening is also correlated with γ‐H2AX foci formation in ageing and cancer cells. Studies have found that triple‐negative breast cancer (TNBC) has shortened telomeres, and γ‐H2AX is exploited as a prognostic biomarker for TNBC [113]. Apart from γ‐H2AX, defective H3S10 and H3S28 phosphorylation also associate with dysregulation of transcription, contributing to carcinogenesis [103, 114].
In the context of cancer therapy, γ‐H2AX can be used to indicate the efficiency of the treatment. Preoperative radiotherapy, a standard therapy for rectal cancer, prevents the local recurrence of the disease. γ‐H2AX is used to correlate with the radiosensitivity of patients with colorectal cancer [115]. Another study indicates its role as the first molecular marker identified that can reveal the survival heterogeneity in γ‐H2AX‐positive breast cancer [116]. Thus, elevated γ‐H2AX can be used both as a marker of cancer and an indicator of cancer therapy, and targeting γ‐H2AX has the potential to be used in combination with other therapies to increase therapeutic efficiency.
Histone acetylation
Histone acetylation is a process by which acetyl groups are added to the lysine residues of histone tails. This post‐translational modification can neutralize the positive charge on the lysine residue, thereby weakening the interactions between histones and DNA and contributing to opening the chromatin, and making it transcriptionally active [117]. Acetylation, which is catalysed by histone acetyl transferases (HAT), increases chromatin accessibility for the RNA polymerase II machinery, DNA repair proteins and other transcription factors [118]. On the contrary, histone deacetylases (HDACs) are involved in the deacetylation of histones post‐repair, restoring the positive charge to the histone and making the chromatin more compact to stabilize the genome.
Histone acetylation in the DDR
Histone acetylation regulates the DDR by detecting the damage, condensing the chromatin to repress transcription, opening chromatin to recruit DNA repair machinery and restoring transcription.
Upon DNA damage, histone acetylation plays a role in chromatin remodelling for efficient DNA repair [119]. Once γ‐H2AX occurs at the damage site, H4 acetylation causes dynamic changes in the chromatin by disrupting the inter‐nucleosomal interactions, resulting in the opening of chromatin, recruiting an optimum level of 53BP1 and enhancing DNA repair [117]. H4 and H2A acetylation mediated by the NuA4‐TIP60 complex is vital to transform the chromatin from its repressive state to an active one, providing a platform for DNA repair. After DNA damage is sensed, NuA4 is recruited through a direct interaction with the MRX complex and it spreads along the DNA resection ends. It has an antagonistic regulatory function on NHEJ factors, favouring an alternative choice of DNA repair pathway [120]. It is shown that NuA4‐TIP60 mutants can cause defective repair and increase genome instability. In addition, the TIP60 complex is involved in recruiting H2AZ, a variant of H2A, modulating gene expression and altering the chromatin structure locally favouring DNA repair [121]. H4K16 acetylation (H4K16ac) by MOF exhibits a biphasic pattern, undergoing deacetylation immediately after damage, followed by acetylation to modulate 53BP1 recruitment. These biphasic dynamics suggest that H4K16ac plays an important role in fine‐tuning the level of 53BP1 after DNA damage [122]. Thus, H4K16ac is a key regulator that controls the choice of the repair pathways through regulating 53BP1 recruitment. Elevated H4K16ac activates ATM and promotes resection that facilitates HR, while lower levels can attenuate resection, proceeding towards NHEJ [123]. Meanwhile, other histone acetylation modifications such as H3K18ac [124, 125] and H2AK15ac [126], favour and promote NHEJ and HR respectively. The immediate γ‐H2AX also orchestrates with other histone acetylation marks. A negative correlation between γ‐H2AX and the level of H3K9ac and H3K56ac was observed [127]. Since γ‐H2AX upon damage recruits a HAT enzyme, KAT2A, it can be hypothesized that γ‐H2AX could indirectly influence the restoration of H3K9ac and H3K56ac marks. This suggests that γ‐H2AX ensures transcription repression at the damaged sites, followed by chromatin opening mediated by H4K16ac, the recruitment of DNA repair proteins, and H3K56 hypoacetylation to promote NHEJ [128].
Once the chromatin opens, H3K14ac allows the retention of the chromatin structure remodelling complex (RSC) and enhances the removal of cyclobutane pyrimidine dimer (CPD) by DNA repair proteins [129]. Evidence suggests that histone chaperone anti‐silencing factor 1(ASF1) can lead to a crosstalk between H3K14ac and H3K56ac [130]. ASF1‐bound H3 can better serve as a substrate for GCN5‐mediated H3K14ac and further acetylate H3K56 [130]. Atomistic molecular dynamics simulations show how H3K56ac exposes DNA damage sites for lesion sensing, making it accessible to DNA repair proteins [131]. Loss of H3K14ac exhibits a decrease in the level of H3K56ac, causing genome instability [130].
After the repair process is completed, restoring the chromatin is essential to maintain its compact and condensed state, which can otherwise lead to cancer, neurodegeneration and ageing [132]. HDAC1 and HDAC‐2 are recruited to the DSB sites after repair is completed, where they remove the H3 and H4 acetylation marks [133]. H4K16ac is removed to the baseline to maintain the chromatin structure. In yeast and mammals, H3K56ac is removed to facilitate assembling new nucleosomes after repair, stabilizing the chromatin [134, 135].
Histone acetylation in transcriptional regulation during the DDR
While histone acetylation‐mediated chromatin relaxation is essential for allowing DNA repair proteins' access to lesion sites, the resulting open chromatin state can create a conflict between transcription and repair machineries. This interference may compromise repair fidelity or generate aberrant transcripts. Therefore, beyond its chromatin remodelling function, histone acetylation also plays a critical role in establishing and maintaining transcriptional repression during the DDR.
As the first step of the DDR, loss of certain histone acetylation marks on the DNA damage site triggers transcription repression. [136]. H3K9ac marks, under untreated conditions, provide active transcription. Upon DNA damage, ATM activation phosphorylates H2AX, MDC1 and KAP1 (a transcriptional co‐repressor) [137]. Phosphorylation of KAP1 on serine824 recruits the NuRD complex consisting of HDAC1and HDAC2 and causes deacetylation of H3K9 and transcription silencing at the damage site [137, 138, 139]. Another study indicates that loss of H3K27ac, an active chromatin mark, immediately after micro‐irradiation at the DSB site, contributes to silencing transcription [140]. To facilitate faithful DNA repair, it is essential to prevent the collision of DNA repair and premature transcripts. Hence, it is essential to regulate premature transcription termination near DNA lesions. In yeast, H3K4me3 promotes recruitment of Nrd1, responsible for efficient termination in a Set1‐dependent manner [141]. While loss of Set1 correlates with increased acetylation and termination defects, the role of histone acetylation in efficient regulation of transcription termination can be speculated.
Furthermore, to facilitate DNA repair, histone acetylation marks can regulate transcription initiation, elongation and termination processes of DNA repair genes. In response to p53 activation, H3 and H4 acetylation on the promoters reshapes the chromatin and releases RNA Pol II from the promoter, promoting the transition from transcription initiation to elongation [142, 143]. H4K16ac deposition by MOF elevates the expression of repair proteins, through recruiting readers such as bromodomain‐containing protein 4 (BRD4). BRD4 can recruit the p‐TEFb complex, which phosphorylates RNA Pol II CTD at serine 2 and leads to the release of paused Pol II, allowing it to proceed to elongation [144]. BRD4 is also a master regulator of transcriptional recovery through re‐engaging RNA Pol II. This suggests that H4K16ac can regulate transcription activation of repair genes and restore recovery of transcription post‐repair. Post‐repair, H3K9ac and H3K27ac are restored and recruit the TFIID subunit of the RNA Pol II pre‐initiation complex to the gene promoters, thereby restoring the initiation of transcription [145].
In the context of diseases, H3K27 hyperacetylation decreases the activity of HDACs, sirtuins, as well as causing transcriptional dysregulation during Parkinson's disease pathogenesis [146]. Defects in various subfamilies of HATs like CREBBP, EP300, KAT6A, KAT6B and histone deacetylase HDAC4 cause several disorders such as Rubinstein–Taybi Type 1 and Type 2 syndromes, KAT6A syndrome, genitopatellar syndrome, Say–Barber–Biesecker–Young–Simpson syndrome and brachydactyly mental retardation syndrome [147], respectively. They are mostly congenital diseases that are caused due to dysregulated transcription. In the context of DNA damage‐related diseases, dysregulated histone acetylation patterns, primarily contribute to cancer, by leading to aberrant expression of genes involved in the cell cycle, apoptosis and tumour differentiation. Studies indicate that an increase in H3K4ac in promoter regions of EMT marker genes, involved in the hedgehog signalling pathway, facilitates cancer cell migration and invasion in head and neck cancer [148]. Another study shows a negative correlation of SIRT1 and H3K4ac in breast cancer cell lines. Thus, HDAC1 can be targeted to affect SIRT1 function as a therapy for breast cancer [149]. Sirtuins, a well‐known protein family pertaining to class III HDACs, is utilized as a therapeutic in ageing and longevity. Reports suggest that caloric restriction activates sirtuins, reducing histone acetylation and increasing lifespan in mice, deacetylating H4K16ac at pro‐apoptotic genes and reducing inflammation as well as acting as a tumour suppressor [150, 151]. Hence, this indicates the possibility of regulating histone acetylation markers towards therapeutic approaches.
Histone methylation
Histone methylation is the addition of one, two, or three methyl groups to lysine or arginine residues in histone tails [152]. Methylation occurs in both heterochromatic and euchromatic regions and is involved in transcription activation, transcription repression and DNA repair [153]. Methyl groups are added by specific histone lysine methyltransferases (KMTs), with S‐adenosylmethionine (SAM) acting as a methyl group donor [154], and removed by histone demethylases (KDMs). Although histone methylation cannot directly alter the electrostatic property of chromatin, it can influence how proteins interact with it through recruiting various readers that recognize this modification; therefore, it plays a crucial role throughout the DDR process.
Histone methylation in the DDR
Unlike histone acetylation, which is generally involved in the opening of chromatin and transcriptional activation, histone methylation plays a dual role in transcription regulation. Its effects depend on both the specific modified residue and the methylation states, enabling either active or repressive chromatin states. Due to its functional variability, histone methylation is involved in multiple DDR processes, including recognizing DNA damage, opening chromatin, recruiting DNA repair proteins and restoring post‐repair transcription. Dysregulation of histone methylation leads to genome instability, cancer and other age‐related disorders.
H3K4me3 is an active transcription mark. Immediately following DNA damage, demethylation of H3K4me3 by KDM5B is essential for repressing local transcription and compacting chromatin by recruiting the zinc finger MYND‐type containing 8 (ZMYND8)‐NuRD chromatin remodelling complex. H3K4me3 on γ‐H2AX sites can impair the recruitment of DNA repair proteins [155], and loss of KDM5B prevents activation of both NHEJ and HR repair pathways [156]. Thus, H3K4me3 is one of the first events post DNA damage, in yeast and mammals, accumulating at the DSB sites [144]. Later on, H3K4me3 is increased on specific cell cycle genes to induce cell cycle arrest [157]. In addition, a study in C. elegans shows that H3K4me2 deposition is increased upon the completion of DNA repair and regulates the recovery of transcription and protein biosynthesis (a topic that will be further discussed in the next section) [158]. In mammals, loss of LSD1, a demethylase enzyme for H3K4me1/2, can promote HR in response to DSBs [159, 160]. These studies show how different states of methylation on the same amino acid residue exhibit different functions during the DDR. Thus, further investigation on how H3K4 methylation and the HMT, MLL‐COMPASS complex are regulated in DDR is required.
Apart from H3K4 methylation, other histone methylations exert different functions during the DDR. H3K9 methylation protects genome integrity by maintaining the heterochromatin organization [161] without compromising DNA repair efficiency. Heterochromatin presents a physical barrier to the DNA repair machinery; thus, demethylation of H3K9 is a crucial step for ensuring efficient DNA repair. Loss of KDM4B, which is responsible for demethylating H3K9me3 and H3K9me2, can increase persistent γ‐H2AX, suggesting KDM4B acts as a DDR protein to enhance repair and confer resistance after radiation [62]. One of the most understudied histone methylation marks is H3K79me, which occurs in the globular domain of H3, rather than its histone tail. Upon damage, 53BP1 recognizes DOT1L‐mediated H3K79me2 via its Tudor domain and then blocks resection to promote NHEJ [162, 163]. It seems that H3K79me2 is involved in choosing the repair pathway; however, whether removal of H3K79me2 promotes the engagement of an error‐free repair pathway is not yet studied. Also, H4K20 methylation plays a fundamental role in the DNA repair process. SETD8 is required to monomethylate H4K20, which is further methylated by SUV4‐20H1 and SUV4‐20H2 leading to di‐ or trimethylation. After UV irradiation, SET8 is degraded by CRL4 ubiquitin ligase in a PCNA‐dependent manner, resulting in the degradation of H4K20me1 to control cell proliferation [164]. An alternate pathway to regulate DNA repair by H4K20 methylation is by recruiting 53BP1. Upon damage, the tandem Tudor domain of 53BP1 recognizes H4K20me2 favouring NHEJ. H2AK15ub is important to recruit 53BP1 upon damage, and thus, the dual interaction with these histone marks helps in the retention of 53BP1 at the damage foci, contributing to cell cycle arrest at the G2/M phase [165]. The crosstalk between H3K79 methylation and H4K20 methylation to recruit 53BP1 and whether loss of H4K20 methylation exhibits a bias towards HR have not been investigated.
Histone methylation regulating transcription during the DDR
Histone methylation serves as a dual‐function chromatin mark that either represses transcription upon damage or promotes initiation and elongation of transcription during and post DNA repair. Upon DNA damage, TIP60 acetyltransferase, mediated by ATM signalling, allows access of SUV39H1 to deposit H3K9me2 and H3K9me3 at the DSB sites. These marks can recruit HP1 and cause chromatin compaction, making the RNA Pol II machinery dissociate from the promoters, leading to transcription repression at the break [166]. Another way to silence transcription locally at the damage site is mediated by polycomb repressive complex (PRC). Enhancer of zeste homologue (EZH2), a subunit of PRC2, can deposit H3K27me3 on the transcribing regions of the damage sites and silence the ongoing transcription, providing time for DNA repair [167]. However, whether H3K9me2/3 and H3K27me3 complement each other to repress transcription is not yet studied.
During the next phase of DDR, which is DNA repair, the transient deposition of the transcription silencing marks is removed, to maintain the open chromatin structure and facilitate DNA repair. KAP1, upon damage acts as a scaffold protein, and interacts with HP1 and CHD3. ATM activation upon DNA damage phosphorylates KAP1 at serine 824, which in turn opens the heterochromatin architecture and temporarily loosens the chromatin for repair [168]. Loss of KDM4A demethylase for H3K9me3 is proved to show a delay in transcription restoration post‐repair. Simultaneously, KDM6A erases H3K27me3, enabling the binding of TFIID to the RNA Pol II and the promoters, and facilitates transcription recovery post‐repair [95, 169]. Loss of both repressive marks can prevent transcriptional silencing after the completion of repair.
Along with the removal of the repressive marks, it is essential to re‐establish the active histone methylation marks, to regulate transcriptional recovery. H3K9 demethylation provides a transcription‐friendly chromatin environment, facilitating MLL‐COMPASS to deposit H3K4me3 and restore transcription. In yeast, it has been shown that H3K4me3 is recruited at sites of DSBs and facilitates repair, allowing for transcriptional recovery [144]. Removal of H3K9 methylation upregulates the expression of DNA repair factors such as BRCA1, and RAD51 for HR while it could also be one of the required factors for the methylation of H3K4 [170]. A further mechanism to elucidate the crosstalk between the repressive H3K9 methylation and the active H3K4 methylation in the context of post‐repair transcription recovery can be investigated. In addition, in C. elegans, deposition of H3K4me2 is observed at 24 h post DNA damage at the gene bodies [158], and it is correlated with transcriptional recovery without affecting the DNA repair process. This creates the intriguing question of whether H3K4me2 is a general marker for transcriptional recovery in response to transcription stress without DNA damage.
H3K36me3 is associated with transcription elongation and is generally deposited on the 3′ ends of genes. Chromatin opening mediated by ATM/ATR signalling re‐engages SET1D to deposit H3K36me3 [171, 172]. This mark along the gene bodies, further recruits SPT16, causing serine 2 phosphorylation at the CTD of RNA Pol II and stabilizing the elongation complex, hence resuming transcription elongation specifically post‐repair [172, 173]. However, there are no reports about the crosstalk between H3K36me3 and repressive marks such as H3K9me and H3K27me, so whether the loss of demethylation of H3K9 and H3K27 affects the levels of H3K36me3 during transcription recovery is unclear.
Dysregulation of histone methylation marks plays a vital role in various ageing‐related disorders. In Alzheimer's disease, H3K9me3 is increased in the temporal cortex and hippocampus, involved in the functions of synaptic plasticity. This causes heterochromatin formation and silencing of gene expression and transcriptional repression [174]. Similarly, altered H3K27me3 marks can contribute to uncontrolled cell proliferation and metastasis, leading to cancer. Hypermethylation of H3K27me3 is observed in colorectal cancer, pancreatic cancer and prostate cancer [175]. H3K4me3 is increased on genes that determine haematopoietic stem cell (HSC) identity, affecting the functions of HSC during ageing [176]. Sarcopenia, an age‐related disorder causing a decline in muscle mass, is found to have altered changes in H3K4me3 [177]. Moreover, H3K4me and its role in regulating lifespan are well conserved among species. Loss of H3K4me3 can extend lifespan in a germline‐dependent manner in C. elegans. In mice, caloric restriction reduces H3K4me3 at the promoters of inflammatory genes, increasing its health span. Inhibition of H3K4 methylation also extends lifespan in yeast [178]. The above studies, highlighting the dynamics of histone methylation marks and their impact in ageing and longevity, show how these chromatin marks can be exploited to control organism ageing and functional decline in the DDR.
Histone ubiquitination
Histone ubiquitination represents a distinct form of post‐translational modification, characterized by the attachment of ubiquitin, a 76 amino acid protein, to lysine residues on the target protein via an isopeptide bond [179]. This modification is regulated by three major ubiquitin enzymes: E1 (ubiquitin‐activating enzyme), E2 (ubiquitin‐conjugating enzymes) and E3 (ubiquitin ligases). E1 catalyses ATP‐dependent ubiquitin activation and creates a thioester bond between the ubiquitin C terminus and the catalytic cysteine on E1. Ubiquitin is then transferred to the catalytic cysteine on E2 and deposited to the target protein by E3 [180]. Protein ubiquitination is majorly known for its role during proteasome‐mediated degradation of targeted proteins. However, histone ubiquitination (mainly mono‐ubiquitination) is involved in the DDR primarily in a proteasome‐independent manner. Deubiquitination is achieved through the activity of deubiquitinating enzymes (DUBs) that interact with the ubiquitinated proteins and act as proteases to break the isopeptide bond binding ubiquitin to the target protein, thereby regulating biochemical activities on the chromatin.
Histone ubiquitination in the DDR
Histones are the most abundant proteins that are ubiquitinated, majorly on the lysine residues of H2A, H2B and H4. Upon DNA damage, RNF8, an E3 ubiquitin ligase, is recruited to the damage sites by ATM phosphorylation and ubiquitinates linker histone H1 [181]. Later, this initial ubiquitination causes the recruitment of RNF168 E3 ligase, which in turn binds to the K63‐linked ubiquitin chains and catalyses mono‐ubiquitination of H2AK13 and K15. These modifications can recruit repair proteins such as 53BP1 (favours NHEJ) and the BRCA1‐A complex (favours HR) [182]. Defects in the H2AK13 and K15 ubiquitination lead to the loss of genome integrity and can cause an immunodeficiency syndrome called the RIDDLE syndrome [183]. RNF8 and RNF168 can also ubiquitinate H2AK127, which recruits SMARCAD1 in an ATP‐dependent manner and promotes DNA end resection, favouring HR [184].
DNA damage‐induced ATM activation also recruits the PRC1 complex consisting of the RING1B E3 ligase, that can ubiquitinate H2A on K119. These modifications are important for condensing the chromatin and preventing the binding of transcription factors, repressing transcription [185]. In addition, H3 and H4 ubiquitination mediated by CUL4‐DDB‐ROC1 is present in UV‐damaged DNA, recruiting XPC‐RAD23Bthat repair thymine dimers stabilizing the genome [186].
After the repair process is complete, the BRCC36 subunit of the BRCA1‐A complex acts as a K63 chain‐specific DUB to remove the ubiquitin groups from the chromatin [187].
In summary, histone ubiquitination is vital for remodelling the chromatin, regulating transcription, recruiting repair machinery and deciding repair pathway choice during the DDR.
Histone ubiquitination in transcriptional regulation during the DDR
DNA damage can cause an imbalance of transcription and repair machinery. At DSB sites, H2AK13 and H2AK15 ubiquitination can recruit the PRC1 subunit RING1B that further ubiquitinates H2AK119. This mark can stabilize the silenced chromatin to prevent inefficient repair by recruiting PRC2 to deposit H3K27me3, a repressive transcription mark [188, 189].
Another ubiquitination mark, H2BK120, is deposited by RNF20, and plays a role in activating transcription during the DDR. Upon DNA damage, H2BK120ub1 opens the chromatin and recruits NHEJ and HR repair factors such as XRCC4, BRCA1 and RAD51, facilitating DNA repair [190]. It can promote the accumulation of the SPT16 subunit of the FACT complex, allowing RNA Pol II to move across gene bodies, promoting transcription of genes involved in DNA repair and cell cycle regulation [191]. Furthermore, it can stimulate the DOT1L methyltransferase enzyme, that methylates H3K79 and activates transcription [159, 192]. Evidence suggests that, upon DNA damage, H2BK120 ubiquitination cannot contribute to genome stability on its own, but that it recruits the FACT complex facilitating gene transcription and promoting Pol II‐mediated transcription elongation of SSRP1‐activated genes (which are subunits of the FACT complex) in mouse embryonic stem cells and yeast cells [193]. Studies also show that it can recruit the H3K4me3 enzyme COMPASS complex by Cps35, indicating the presence of crosstalk between H2B ubiquitination and transcriptionally active marks [194]. Further studies can be performed to investigate the role of Cps35 and the FACT complex with other histone modifications and understanding its common function in the transcription process. Ubiquitination on H2BK123 in yeast and mammalian cells is fundamental for DNA damage checkpoint activation. The absence of this mark inhibits RAD9 phosphorylation, allowing DNA repair by halting cell cycle progression and directly influencing RNA Pol II transcription [195].
To restore transcription after DNA repair, USP16, a DUB enzyme, removes the H2AK119ub mark and displaces the PRC components, thus reversing the chromatin repressive state and recovering transcription elongation [196].
Reports show that dysregulated histone ubiquitination in response to DSBs leads to cellular senescence, impaired proteostasis, ageing and neurodegenerative disorders [197]. A study shows that elevated levels of H2A ubiquitination can cause accelerated neurodegeneration and Alzheimer's disease [197]. Reduction of H2Aub increases the adult lifespan in Drosophila melanogaster and is evolutionarily conserved in mice, monkeys and humans [198]. This identifies H2Aub as a universal biomarker of ageing, and its interaction with H3K4 and H3K79 methylations can be investigated in vivo.
Conclusion and future perspective
Transcriptional regulation in response to DNA damage is a highly coordinated process that can be divided into three steps: (1) rapid transcriptional repression at both local and global levels, (2) selective activation of DDR genes, and (3) eventual transcriptional recovery (Table 1). While the involvement of specific histone modifications in each phase has been delineated above, the precise mechanisms governing their dynamic crosstalk remain incompletely understood. A critical unresolved question is how transcription transitions through ‘access‐repair‐restore’ [199] chromatin states in a spatiotemporally controlled manner following DNA damage. For instance, both transcriptional repressive (e.g., H3K9 methylation) and activating (e.g., H3K4 methylation) marks play roles during the DDR. However, the way in which these two opposing marks interact with each other to influence the transcriptional state remains unclear. Key unknowns include how PTM‐modifying enzymes are recruited in response to TBLs and how they coordinate with the transcriptional machinery to regulate gene expression.
Furthermore, current technical limitations hinder the mapping of DNA lesions within high‐order chromatin structures, leaving a gap in our understanding of how chromatin architecture influences transcriptional regulation during the DDR. Another intriguing, yet underexplored aspect is the heritable nature of chromatin modifications. While histone marks are transiently altered during DDR, emerging evidence suggests they may contribute to an ‘epigenetic memory’ of cellular stress, [200] potentially influencing longevity and cellular fitness. Whether these modifications exert long‐term or even transgenerational effects remains an open question. Elucidating these mechanisms could uncover novel therapeutic strategies to mitigate the long‐term consequences of DNA damage.
Given the central role of DNA damage in ageing and cancer, histone modifications present a promising target for therapeutic intervention. For example, SPR5 (H3K4me2 demethylase) inhibition has the potential to modulate lifespan and transcriptional recovery upon UV‐induced damage in a model organism [163]. However, further study is needed to optimize their application—including timing, dosage and tissue specificity—while minimizing off‐target effects. A deeper understanding of histone modification dynamics during the DDR will not only advance fundamental biology but also pave the way for innovative treatments for age‐related diseases and cancer.
Author contributions
AJK wrote the initial draft of the manuscript. SW supervised and revised the final version of the manuscript. All authors read and approved the submitted version.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Mariño‐Ramírez L , Kann MG , Shoemaker BA and Landsman D (2005) Histone structure and nucleosome stability. Expert Rev Proteomics 2, 719–729.16209651 10.1586/14789450.2.5.719PMC 1831843 · doi ↗ · pubmed ↗
- 2Kujirai T and Kurumizaka H (2020) Transcription through the nucleosome. Curr Opin Struct Biol 61, 42–49.31790919 10.1016/j.sbi.2019.10.007 · doi ↗ · pubmed ↗
- 3Petesch SJ and Lis JT (2008) Rapid, transcription‐independent loss of nucleosomes over a large chromatin domain at Hsp 70 loci. Cell 134, 74–84.18614012 10.1016/j.cell.2008.05.029PMC 2527511 · doi ↗ · pubmed ↗
- 4Fenley AT , Anandakrishnan R , Kidane YH and Onufriev AV (2018) Modulation of nucleosomal DNA accessibility via charge‐altering post‐translational modifications in histone core. Epigenetics Chromatin 11, 11.29548294 10.1186/s 13072-018-0181-5PMC 5856334 · doi ↗ · pubmed ↗
- 5Bannister AJ and Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21, 381–395.21321607 10.1038/cr.2011.22PMC 3193420 · doi ↗ · pubmed ↗
- 6Malik S and Roeder RG (2023) Regulation of the RNA polymerase II pre‐initiation complex by its associated coactivators. Nat Rev Genet 24, 767–782.37532915 10.1038/s 41576-023-00630-9PMC 11088444 · doi ↗ · pubmed ↗
- 7Gamarra N and Narlikar GJ (2021) Collaboration through chromatin: motors of transcription and chromatin structure. J Mol Biol 433, 166876.33556407 10.1016/j.jmb.2021.166876 PMC 8989640 · doi ↗ · pubmed ↗
- 8Winter GE , Mayer A , Buckley DL , Erb MA , Roderick JE , Vittori S , Reyes JM , di Iulio J , Souza A , Ott CJ et al. (2017) BET Bromodomain proteins function as master transcription elongation factors independent of CDK 9 recruitment. Mol Cell 67, 5–18.28673542 10.1016/j.molcel.2017.06.004PMC 5663500 · doi ↗ · pubmed ↗