Comparative genomic analyses reveal key traits for biocontrol and the promotion of plant growth in Paenibacillus strains
Luciano Nascimento de Almeida, Mirelly Jady Fernandes e Silva, Blenda de Freitas Rodrigues Jesuino, Sumaya Martins Tupy, Jorge Henrique Resende Vieira, Gabriela Amaral Xavier, João Paulo Lopes da Rocha, Osiel Silva Gonçalves, Mateus Ferreira Santana

TL;DR
This study uses genome analysis to show that Paenibacillus bacteria have many genes that help plants grow and fight pests, making them useful for sustainable agriculture.
Contribution
The study identifies key genomic traits in Paenibacillus linked to plant growth promotion and biocontrol through analysis of 428 genomes.
Findings
Paenibacillus genomes contain numerous genes for plant growth promotion and biocontrol traits like phosphate solubilization and antimicrobial compound production.
The open pan-genome and mobile genetic elements suggest horizontal gene transfer contributes to the spread of beneficial traits.
Paenibacillus strains lack virulence factors and antibiotic resistance genes, indicating low pathogenicity and safety for agricultural use.
Abstract
Paenibacillus species have emerged as promising candidates for sustainable agriculture due to their functional versatility in plant growth promotion and biocontrol. We performed a comparative genomic analysis of 428 high-quality Paenibacillus genomes to assess their ecological adaptability and biotechnological potential. The analyzed strains originated from diverse environments, reflecting broad ecological distribution. Functional annotation revealed a widespread occurrence of plant growth-promoting traits (PGPTs), including phosphate and potassium solubilization, siderophore biosynthesis, nitrogen fixation, and phytohormone-related compounds. On average, the genomes contained 249 genes associated with biofertilization, 190 with phytohormone production, 97 linked to bioremediation, and around 322 involved in competitive exclusion. The open pan-genome configuration (b = 0.503) highlights…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
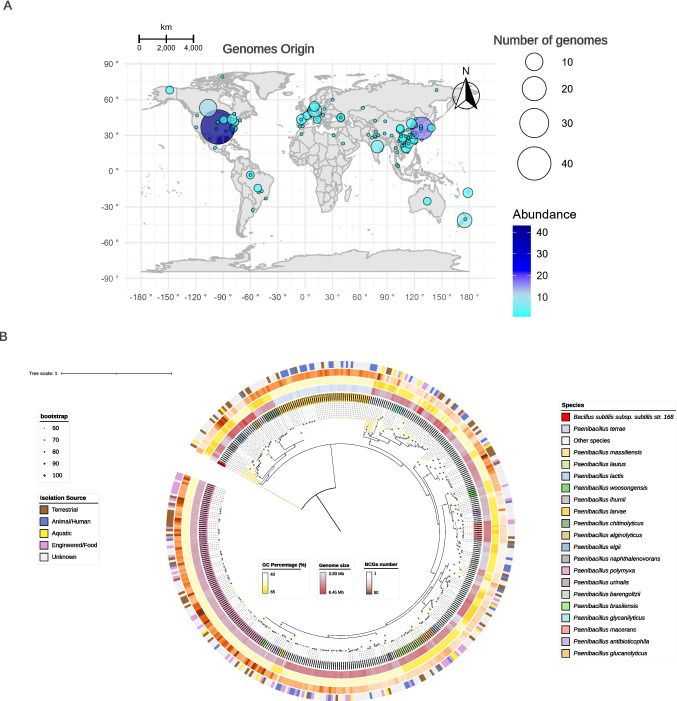 Figure 1
Figure 1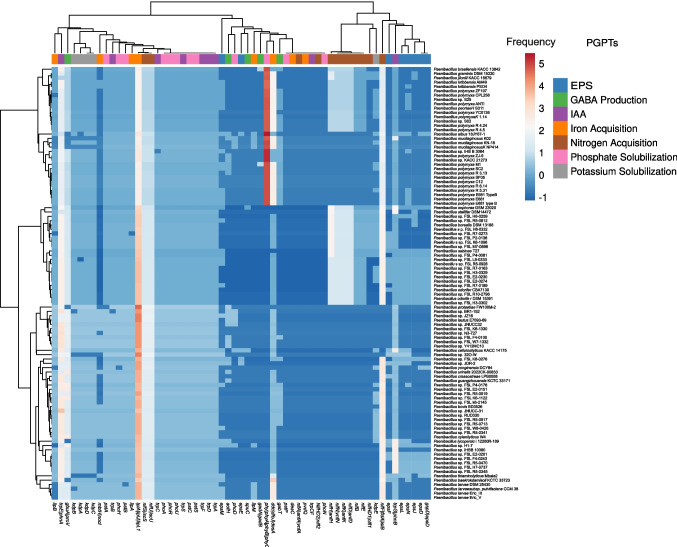 Figure 2
Figure 2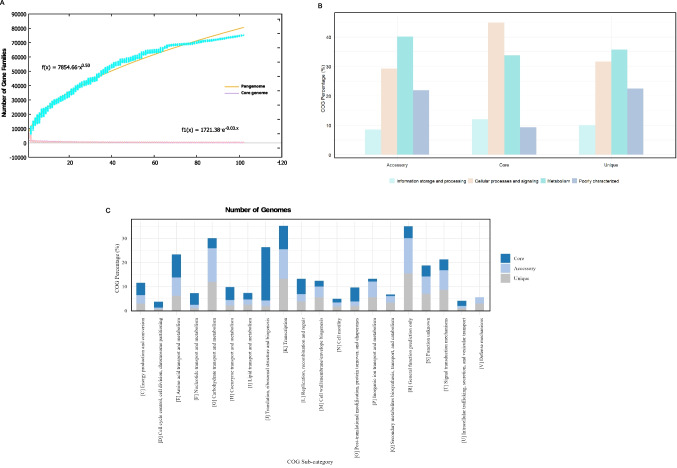 Figure 3
Figure 3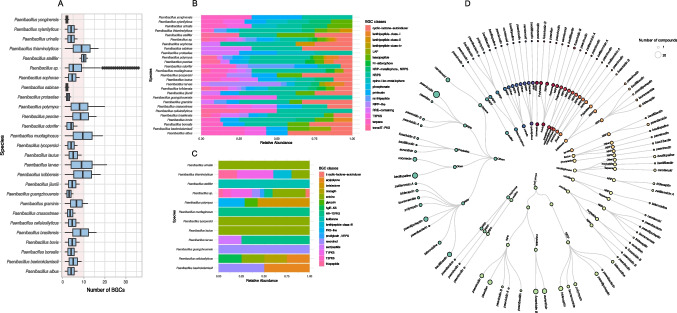 Figure 4
Figure 4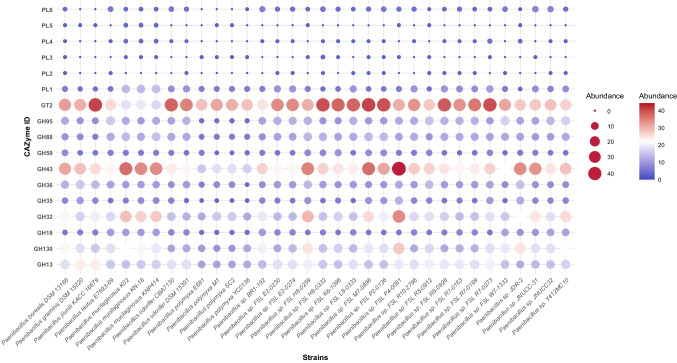 Figure 5
Figure 5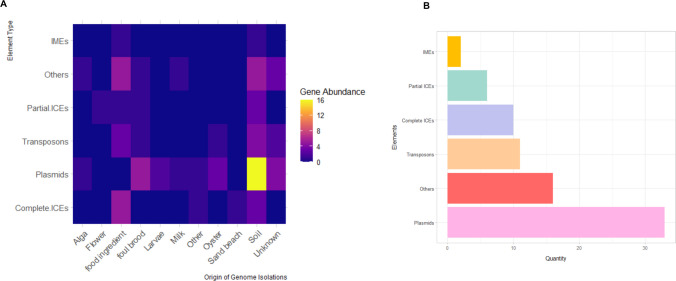 Figure 6
Figure 6- —Universidade Federal De Viçosa
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant-Microbe Interactions and Immunity · Genetic and Environmental Crop Studies · Bacterial biofilms and quorum sensing
Introduction
Paenibacillus is a genus of Gram-positive, facultatively anaerobic, endospore-forming bacteria comprising at least 240 species (Dai et al. 2019; Xie et al. 2016). Initially classified under the genus Bacillus, these bacteria were reclassified as a distinct genus in 1993 following a comprehensive taxonomic reassessment (Ash et al. 1993). Members of Paenibacillus exhibit remarkable biochemical and morphological diversity and inhabit a wide array of environments, including soil (McSpadden Gardener 2004), rhizospheres (Xie et al. 2012), marine sediments (Ravi et al. 2007), clinical specimens (Ouyang et al. 2008), and fermented foods (He et al. 2007), among others. Frequent isolation of Paenibacillus from the rhizosphere, particularly from agriculturally important crops (Raza et al. 2008), underscores the ecological versatility of the genus, reflecting its broad adaptation to diverse environmental niches (Huang et al. 2020).
Over the past two decades, growing attention has been directed towards the ecological significance and biotechnological potential of Paenibacillus species (Lal and Tabacchioni 2009). Numerous species have been recognized as plant growth-promoting rhizobacteria (PGPR), exerting both direct and indirect effects on plant development. Direct mechanisms include the biosynthesis of phytohormones, phosphate solubilization, and biological nitrogen fixation. Indirectly, plant growth is stimulated through the suppression of phytopathogens via the secretion of antimicrobial metabolites and hydrolytic enzymes (Soni et al. 2021). Several studies have demonstrated the efficacy of Paenibacillus species in promoting growth across various crops. For instance, the P. polymyxa CR1 strain has been shown in greenhouse trials to enhance the growth of maize, potato, and cucumber. This strain also exhibited in vitro nitrogen fixation, phosphate solubilization, and production of indole-3-acetic acid (IAA) (Weselowski et al. 2016). Similarly, Paenibacillus sp. YSY-4.3 was found to synthesize phytohormones (IAA, gibberellic acid, and zeatin), form biofilms and siderophores, and exhibit antifungal activity, including chitin degradation (Tran et al. 2024). Furthermore, the P. polymyxa J2-4 strain was recently shown to significantly promote cucumber growth under field conditions (Shi et al. 2024).
Paenibacillus spp. have also demonstrated effectiveness in protecting plants against phytopathogens through diverse biocontrol strategies. These mechanisms have proven effective against a broad spectrum of agriculturally relevant pathogens, including phytopathogenic fungi (Samain et al. 2017; Tran et al. 2024), pathogenic bacteria (Taheri et al. 2022a, b), and plant-parasitic nematodes (Singh and Wesemael 2022a, b; Shi et al. 2024). The production of bioactive secondary metabolites is a hallmark of Paenibacillus strains. Notably, P. polymyxa produces fusaricidin—a non-cationic cyclic lipopeptide with potent antifungal activity—and polymyxin, a cationic cyclic lipopeptide that is highly effective against Gram-negative bacteria (Raza et al. 2008).
Paenibacillus strains have shown strong potential for the biological control of root-knot nematodes belonging to the genus Meloidogyne. A prominent example is the P. polymyxa LMG27872 strain, which exhibited antagonistic activity against Meloidogyne incognita in both in vitro and in vivo experiments, while also promoting tomato growth (Singh and Wesemael 2022a, b). Beneficial and mutualistic rhizobacteria such as Paenibacillus spp., which colonize plant roots, can also trigger induced systemic resistance (ISR)—a defense mechanism that enhances plant immunity in distal tissues, providing broad-spectrum protection against pathogens and pests (Grady et al. 2016). Paenibacillus species have been shown to induce ISR against a wide array of pathogens, including bacteria (Lee et al. 2012; Phi et al. 2010a, 2010b), fungi (Gkizi et al. 2016), nematodes (Khan and Kim 2012), and viruses (Kumar et al. 2016).
To date, numerous Paenibacillus genomes have been sequenced, offering critical insights into the genetic basis of their functional diversity (Huang et al. 2020). Despite these advances, a systematic and comprehensive comparative genomic analysis is still needed to elucidate the genetic determinants underlying plant growth promotion and biocontrol capabilities. Such studies are essential for understanding the prevalence and distribution of these traits, as well as for elucidating the ecological roles of Paenibacillus species particularly within the rhizosphere and in association with plant hosts. In this context, we conducted a comparative genomic analysis of 428 Paenibacillus genomes to investigate the distribution and phylogenetic patterns of plant growth-promoting and biocontrol-related traits. Subsequently, a subset of 97 complete genomes was selected for in-depth functional analyses, enabling the identification of promising candidate species with potential as plant growth promoters and antagonists of phytopathogens. Additionally, we assessed potential risks, including the presence of genes associated with antimicrobial resistance and virulence factors.
Materials and methods
Genomic data collection and Paenibacillus strain distribution analysis
As of August 2024, approximately 1,500 Paenibacillus genome sequences had been deposited in GenBank. From these, 1,345 genomes from the RefSeq database were downloaded for quality assessment using CheckM v1.0.13 (Parks et al. 2015) and BUSCO (Seppey et al. 2019). Genomes meeting the criteria of > 95% completeness and < 0.5% contamination were retained, resulting in a final dataset of 428 high-quality genomes. Among these, 48 genomes belonged to P. polymyxa, while the remaining 380 represented other Paenibacillus species. Metadata regarding the geographic and environmental origins of each strain were retrieved from NCBI and are summarized in (Supplementary Table S1).
Phylogenetic analysis
A phylogenetic reconstruction of the Paenibacillus core genome was performed using the 428 selected genomes. CheckM v1.0.13 (Parks et al. 2015) was employed to extract 43 single-copy universal marker genes using the tree_ga function. These marker protein sequences were concatenated and used for phylogenetic inference via the maximum likelihood method in IQ-TREE v1.6.11 (Minh et al. 2020), applying the best-fit model "LG + I + G4". Phylogenetic tree construction was conducted with the parameters –m TEST –bb 1000 to ensure robustness and statistical support. The resulting tree was visualized and annotated using the Interactive Tree of Life (iTOL) tool (Letunic and Bork 2019).
Identification of plant growth-promoting traits (PGPTs) and biocontrol-associated genes
For functional analyses, only complete genomes were selected, which were annotated using Bakta (Schwengers et al. 2021). Additional details on these genomes are provided in Supplementary Table S2. The comprehensive PGPg_finder pipeline (Pellegrinetti et al. 2024) was used, combining direct sequence annotation with de novo assembly methods to accurately detect the presence of PGPTs in the genomes, encompassing categories of direct and indirect plant growth promotion. The tool cross-references the analyzed sequences with the PlaBAse database (Patz et al. 2021), enabling the identification and quantification of PGPTs in the evaluated genomic datasets. A total of 59 PGPT-related genes, annotated using PgPg_finder and associated with processes such as biofertilization and phytohormone production, were used as indicators of plant-interactive potential (Supplementary Table S3). The frequency of detected genes was transformed using the natural logarithm [ln(x)] and visualized as a heatmap using the online tool ClustVis v1.0 (Metsalu and Vilo 2015). Biosynthetic gene clusters (BGCs) were identified using antiSMASH v7.0 (Blin et al. 2023a, b), while carbohydrate-active enzymes (CAZymes) were annotated through the dbCAN3 pipeline (Zheng et al. 2023), using default detection thresholds.
Pan-genome analysis and functional categorization (COGs)
A subset of 97 Paenibacillus genomes was selected for pan-genome analysis based on genome completeness and assembly quality. Only complete or near-complete genomes with low levels of fragmentation were included in order to minimize annotation artifacts and ensure reliable gene presence/absence inference, which is essential for comparative genomic and functional analyses. This quality-based selection aimed to increase the robustness of the pan-genome analysis. The genomes were annotated using Bakta (Schwengers et al. 2021) and the resulting protein FASTA files were input into the Bacterial Pan Genome Analysis (BPGA) v1.3 pipeline (Chaudhari et al. 2016). This pipeline facilitated clustering into core, accessory, and unique gene families and functional classification according to the Clusters of Orthologous Groups (COG) database. Pan-genome openness was assessed using Heaps’ law, described by the equation n = um·x^b (Tettelin et al. 2008), where x is the number of genomes and um and b are curve-fitting parameters. A value of 0 < b < 1 indicates an open pan-genome, while b < 0 suggests a closed one.
Detection of mobile genetic elements (MGEs)
To investigate mobile genetic elements (MGEs), chromosomal sequences from complete genomes were obtained from NCBI and analyzed using ICEscreen v1.3.2 (Lao et al. 2022) to detect Integrative and Conjugative Elements (ICEs) and Integrative and Mobilizable Elements (IMEs). Only elements with associated signature proteins were considered. ICEs and IMEs were classified into four categories: complete ICEs (possessing recombination, conjugation, stability, integration, and excision modules), complete IMEs (with integration, excision, recombination, and mobilization modules), partial ICEs (with missing components), and other elements (degenerate or unclassified). Transposable elements were identified through BLASTn searches against the TnCentral, INTEGRALL, and ISFinder databases, using thresholds of > 50% coverage and > 70% identity (Ross et al. 2021). Plasmid content was obtained directly from complete genome entries in NCBI. In addition, genes associated with plant growth promotion (PGPTs) and biocontrol, identified using the antiSMASH 7.0, PGPT-Pred, and PGPg_finder tools, were correlated with the geographical locations of isolation and environmental types to assess the environmental influence on the abundance of PGPT genes and secondary metabolites. All identified elements were compared with each other by BLASTn using the Geneious software (https://www.geneious.com) to detect possible gene transfer events. ICEs and IMEs were also compared with the ICEberg 3.0 database (https://tool2-mml.sjtu.edu.cn/ICEberg3/ICEfinder.php) (Wang et al. 2024), being considered similar when they showed coverage greater than 50% and identity above 70%.
Detection of antibiotic resistance genes and virulence factors
To assess the potential risks associated with antimicrobial resistance and virulence, we conducted a comprehensive screening of antibiotic resistance genes (ARGs) and virulence factors (VFs) across all Paenibacillus genomes analyzed. Gene sequence data were queried against multiple curated reference databases. Specifically, ARGs were identified using the Comprehensive Antibiotic Resistance Database (CARD; Alcock et al. 2020), ResFinder (Zankari et al. 2012), MEGARes (Lakin et al. 2017), and ARGANOT (Gupta et al. 2014). Virulence-associated genes were detected using the Virulence Factor Database (VFDB) implemented via the ABRicate tool (https://github.com/tseemann/abricate). Default parameters were used, with a minimum identity cutoff of 80% and minimum coverage threshold of 70%
Data availability
All genomes used in this study are publicly available through the GenBank database (https://www.ncbi.nlm.nih.gov/genCiteESM.bank/.). Supplementary datasets generated and analyzed during this research are provided as supplementary materials accompanying this article.
Results
Paenibacillus strains are widely distributed in a variety of environments, demonstrating high ecological versatility
To investigate the global distribution and ecological range of Paenibacillus species, we compiled a comprehensive dataset from NCBI comprising the 428 high-quality genomes analyzed in this study (Supplementary Table S1). Environmental metadata was used to infer the origin of each strain, revealing considerable heterogeneity in isolation sources. Genomes were derived from diverse habitats, including marine sediments, non-agricultural soils, rhizospheric soils, food ingredients, and seeds, among others. Notably, 13.12% of the genomes were isolated from rhizospheric soils, reinforcing the ecological association of certain Paenibacillus strains with plant hosts.
Geographical analysis showed that the genomes were distributed across multiple continents (Fig. 1A). Genome size among the 428 strains varied widely, ranging from 0.93 Mb to 6.45 Mb, while G + C content spanned from 40.0% to 65.0%. The number of biosynthetic gene clusters (BGCs) identified per genome ranged from 1 to 50. The P. polymyxa clade stood out for harboring a notably high number of BGCs, suggesting strong secondary metabolic potential within this group.Fig. 1. Global distribution and phylogeny of the Paenibacillus. (A) The global distribution of Paenibacillus described in this study is represented on a map generated using the ‘ggplot 2’ package in R, with abundance highlighted by circles of varying sizes (Figure A). (B) Phylogenetic tree constructed using maximum likelihood method with IQ-TREE v.1.6.11, based on 428 Paenibacillus genomes. The analysis utilized 43 single-copy marker genes, identified using CheckM, and was performed under the LG + I + G4 rate heterogeneity model with maximum of 1,000 bootstrap replicates. Bootstrap values are represented by black circles. Each color in the phylogenetic tree corresponds to a clade representing distinct species, as indicated in the legend, including the outgroup Bacillus subtilis. The tree is scaled, with branch lengths in the same units as the evolutionary distances used to infer the phylogenetic tree
To explore evolutionary relationships within the genus, we constructed a robust phylogenetic tree based on the concatenated alignment of 43 universally conserved single-copy marker genes (Fig. 1B). The tree, derived from 428 publicly available RefSeq genomes, exhibited strong bootstrap support for most nodes (> 90%), indicating high phylogenetic reliability. Our analysis resolved 28 well-defined branches, each representing a distinct taxonomic lineage, thereby highlighting the extensive species diversity encompassed within the genus.
Most of the genomes were found to cluster within the P. polymyxa clade, the type species of the genus (Trüper, 2005), reflecting both its ecological prominence and its appeal as a model organism for functional studies. The increasing scientific interest in this species, coupled with advancements and accessibility in sequencing technologies, has led to a notable rise in genomic submissions to public repositories in recent years (Wallner et al. 2024). Other highly represented clades included P. larvae, P. lautus, P. macerans, and P. chitinolyticus, each contributing to the overall genomic and functional diversity observed across the dataset.
Paenibacillus genomes exhibit diverse genetic repertoires for plant growth-promoting traits (PGPTs)
To identify genes associated with plant growth-promoting traits (PGPTs), we analyzed 97 complete Paenibacillus genomes by screening their predicted protein sequences. The PGPTs were classified into broad functional categories involving direct and indirect mechanisms of plant growth promotion. Among the indirect categories, genes related to the colonization of plant-derived substrates and abiotic stress mitigation were particularly prominent. In the direct categories, the most notable mechanisms included iron acquisition, plant vitamin biosynthesis, and phosphate solubilization (Supplementary Fig. 1). On average, the genomes contained approximately 249 genes related to biofertilization, around 190 genes associated with phytohormone production, 97 genes involved in bioremediation, and approximately 322 genes linked to competitive exclusion. These categories represent the major functional groups of plant growth-promoting traits (PGPTs) identified across the analyzed genomes.
Species such as P. mucilaginosus (987 genes), P. thiaminolyticus (920 genes), and P. borealis (919 genes) exhibited the highest total numbers of PGPT-related genes, particularly within the biofertilization category, surpassing even P. polymyxa (850 genes). These findings suggest that these species may play a more significant role in promoting plant growth.
Analysis revealed substantial variation in the frequency and distribution of PGPT-related genes across the analyzed genomes, highlighting the functional diversity and differential plant-associative potential among Paenibacillus species. Strains isolated from soil environments exhibited the highest absolute numbers of genes related to plant growth promotion compared to isolates from other environments, totaling 5,522 genes associated with biofertilization, 2,288 genes related to bioremediation, 4,164 genes linked to phytohormone production, and 7,057 genes related to competitive exclusion. However, when the average number of PGPT-related genes per isolate was calculated for each environment to correct for sampling size bias, isolates from plant-associated environments (900) and the rhizosphere (874) showed higher averages than those from soil (865).
As illustrated in Fig. 2, genes related to phosphate solubilization—phyA (phytase A), phyB (phytase B), and phyC (phytase C)—were among the most frequently detected, occurring up to 14 times in individual genomes. These genes were predominantly found in P. polymyxa, P. kribbensis, P. brasilensis, and P. graminis. Additional phosphate metabolism genes, including phoA (alkaline phosphatase), phoD (alkaline phosphatase D), phoH (phosphate starvation-inducible protein), and phoR (phosphate regulon sensor kinase), were less abundant, although phoA showed a consistent presence across all analyzed genomes.Fig. 2A heatmap describing the distribution of genes associated with plant growth-promoting traits (PGPTs) that directly contribute to plant growth. The abundance values were log-transformed using ln(x). Row centering and unit variance scaling were applied to improve visualization. Missing values were estimated using imputation techniques. Rows and columns were clustered using correlation distance and average linkage methods
Siderophore-associated genes, particularly dfoJ (desferrioxamine biosynthesis protein) and desA (desferrioxamine biosynthesis enzyme), were detected in all genomes, with frequencies ranging from two to eight copies per genome, suggesting a conserved genetic potential for iron acquisition. Genes associated with indole-3-acetic acid (IAA) biosynthesis — including trpA (tryptophan synthase alpha subunit), trpB (tryptophan synthase beta subunit), trpC (indole-3-glycerol phosphate synthase), trpD (anthranilate phosphoribosyltransferase), trpE (anthranilate synthase component I), iaaM (tryptophan monooxygenase), iaaH (indole-3-acetamide hydrolase), ipdC (indole-3-pyruvate decarboxylase), and ppdC (phenylpyruvate decarboxylase) — were detected across several Paenibacillus genomes (Supplementary Table S3). These genes are mainly associated with tryptophan-dependent auxin biosynthesis pathways, particularly the indole-3-pyruvate (IPyA) pathway, which is widely reported in plant-associated bacteria. Although auxin-related genes were broadly distributed, differences in gene composition and copy number were observed among species, indicating variability in the metabolic potential for auxin biosynthesis within the genus (Supplementary Table S3). The presence of these genes suggests a genetic potential for auxin biosynthesis within the genus, although the completeness of individual pathways requires further investigation.
Regarding γ-aminobutyric acid (GABA) production, the genes gbuA (GABA permease subunit) and proV (glycine betaine/proline ABC transporter substrate-binding protein) were widely distributed, appearing in most genomes with frequencies between four and six copies. Genes associated with potassium solubilization—kdpA (potassium-transporting ATPase subunit A), kdpB (subunit B), kdpC (subunit C), and kdpD (sensor histidine kinase)—were present in most genomes at a frequency of approximately two copies each. More than 20 Paenibacillus species are known to fix atmospheric nitrogen, and the nitrogen fixation (nif) gene cluster remains highly conserved among these nitrogen-fixing strains (Xie et al. 2014). In our dataset, nine key genes involved in nitrogen fixation—nifA (transcriptional regulator), nifB (FeMo-cofactor biosynthesis protein), nifD (nitrogenase alpha subunit), nifK (nitrogenase beta subunit), nifS (cysteine desulfurase), nifU (Fe–S cluster scaffold protein), nifH (nitrogenase reductase), nifF (ferredoxin), and nifN (FeMo-cofactor biosynthesis protein)—were identified. Among these, nifS and nifU were ubiquitous across all analyzed genomes, whereas nifK, nifD, and nifH were present in approximately 38% of the genomes, generally with lower copy numbers (two to six). The species with the highest representation of nitrogen fixation-related genes included P. polymyxa, P. jilunlii, P. sabinae, and P. brasiliensis. Finally, genes related to exopolysaccharide (EPS) biosynthesis—epsL (glycosyltransferase), epsJ (EPS biosynthesis protein), epsE (priming glycosyltransferase), and epsM (EPS export protein)—were detected at low frequencies, with most genomes containing no more than two copies. Despite their limited abundance, the presence of these genes suggests a potential for biofilm formation and enhanced root colonization in certain strains.
The Paenibacillus pan-genome is open and functionally diverse across strains
The pan-genome analysis of Paenibacillus strains revealed a total of over 80,000 gene families, with an average of 418 core genes, 4,631 accessory genes, and 355 unique genes per genome. Core genes accounted for approximately 7.73% of each genome, while accessory and unique genes represented, on average, 85.7% and 6.57%, respectively. According to Heaps’ Law (b = 0.503), the Paenibacillus pan-genome remains open and expanding, reflecting high genomic plasticity and the continuous acquisition of new genes (Fig. 3A). Notably, species such as P. thiaminolyticus, P. cellulosilyticus, and P. baekrokdamisoli exhibited the highest proportions of unique, species-specific genes, suggesting specialized ecological roles.Fig. 3. Graphic representation of the pan-genome and core-genome of 97 complete Paenibacillus genomes. (A) The plot shows that the equations fit the total and core genome families, as well as how the number of gene families increases and decreases in the pan-genome and core-genomes as more genomes are added. (B) Displays the functional proportions of core, accessory, and unique genes in the COG categories. (C) Distribution of COG subcategories
Functional classification based on Clusters of Orthologous Groups (COG) demonstrated distinct patterns among gene categories (Fig. 3B). The core genome was primarily enriched in genes associated with cellular processes and signaling (44.86%), particularly within class J (22.28%; translation, ribosomal structure and biogenesis), class E (9.63%; amino acid transport and metabolism), and class C (5.11%; energy production and conversion). In contrast, these same functional classes were proportionally reduced in the accessory (J: 2.31%, E: 7.54%, C: 3.49%) and unique genomes (J: 1.89%, E: 6.23%, C: 2.96%). Meanwhile, the accessory and unique gene fractions were strongly enriched in functions associated with environmental adaptation and defense, including class K (12.24% and 13.28%; transcription), class G (13.98% and 11.95%; carbohydrate transport and metabolism), class T (8.10% and 8.71%; signal transduction), class Q (2.77% and 3.35%; secondary metabolite biosynthesis, transport, and catabolism), and class V (2.63% and 2.95%; defense mechanisms) (Fig. 3C). Collectively, these findings indicate that while the core genome retains genes essential for fundamental cellular maintenance, the accessory and unique genomes provide the functional versatility necessary for environmental resilience, host interaction, and niche specialization across diverse Paenibacillus strains.
The broad variability of biosynthetic genes in Paenibacillus lineages suggests the existence of a shared metabolic core
Analyses performed with AntiSMASH identified numerous secondary metabolite clusters across the Paenibacillus genomes. A total of 1,064 Biosynthetic Gene Clusters (BGCs) were identified and distributed into 163 different types within our dataset. The most frequent BGC was the NRP (Non-Ribosomal Peptides), with 130 occurrences, representing 100% of the maximum frequency. Other prominent clusters included RiPP (Ribosomally Synthesized and Post-Translationally Modified Peptides) with 85 occurrences (65.38%) and NRP + Polyketide with 55 occurrences (42.31%). Less frequent clusters, such as NRP: Glycopeptide and Polyketide: Iterative type I polyketide + Saccharide, were observed only once each, representing 0.77% of the total. In addition, a total of 128 unique metabolite types were identified, reflecting the significant diversity of compounds produced by the genus Paenibacillus. The strains with the largest biosynthetic regions were P. larvae subsp. larvae strain Eric_III (21 regions) and P. polymyxa strain R 3.13 (20 regions) (Supplementary Table S5).
A comparative analysis of the number of BGCs identified across different Paenibacillus species revealed substantial variability, with an average of 9.5 BGCs per genome (Fig. 4A). The average number of BGCs per species varied considerably, with P. larvae, P. polymyxa, and P. mucilaginosus exhibiting the highest averages (20, 18, and 17, respectively), while species such as P. sabinae and P. yonginensis showed lower numbers. Notably, Paenibacillus sp. exhibited a broad dispersion in the number of BGCs (Fig. 4A), indicating significant genomic diversity within this unidentified group. This pattern may reflect the inclusion of heterogeneous genomes or differences in environmental conditions at the time of isolation.Fig. 4. Abundance and diversity of biosynthetic gene clusters (BGCs) and their associated secondary metabolites across species of the genus Paenibacillus. A. Boxplot showing the distribution of the number of BGCs across Paenibacillus species. The red highlight indicates the average of 9.5 BGCs per genome. B. Relative abundance of core BGC classes across different Paenibacillus species. C. Relative abundance of exclusive BGC classes across different Paenibacillus species. Circle size represents the normalized number of a specific compound. D. Distribution of bioactive secondary metabolite compounds within Paenibacillus species. Additional details are available at Supplementary Table S5
The presence of conserved BGCs across Paenibacillus species suggests the existence of a shared metabolic core (Fig. 4B). Classes such as transAT − PKS, T3PKS, proteusin, NRPS, lanthipeptide-class-I, lanthipeptide-class-II, and terpenes were widely distributed among species, representing a significant fraction of the BGCs in nearly all the strains analyzed. In contrast, several species exhibited biosynthetic classes that were either exclusive or rarely shared (Fig. 4C). These specific classes are indicative of metabolic specializations and may be linked to ecological adaptations or unique environmental interactions. Species such as P. polymyxa and P. mucilaginosus featured classes like glycoin and betalactone, which were not widely represented in other species. P. baekrokdamisoli and P. guangzhouensis showed high proportions of resorcinol (Fig. 4C). Other classes, such as thiopetide, prodigiosin, ectoine, and crocagin, were confined to a small set of species, underscoring their functional exclusivity.
Finally, we gained insights into the distribution of key bioactive compounds within the genus (Fig. 4D). Analysis of our dataset revealed that a considerable portion of well-characterized secondary metabolite clusters with potential biocontrol activity exhibited similarity greater than 70%. The most abundant BGCs included Bacillopaline, Tridecaptin, Fusaricidin B, Bacillibactin, Paeninodin, and Polymyxin, with Bacillopaline (24), Tridecaptin (22), and Fusaricidin B (22) being the most prominent. In total, 186 clusters with similarity > 70% were identified. The results indicate that P. polymyxa strains harbor the highest number of well-characterized BGCs for biological control, with an average of seven clusters per genome. In addition to P. polymyxa, other strains, such as Paenibacillus thiaminolyticus Mbale2, Paenibacillus cellulosilyticus, and P. cellulosilyticus KACC 14175, also presented a considerable number of BGCs, including Polymyxin, Ectoine, Paeninodin, Fusaricidin B, and Tridecaptin. Furthermore, our findings suggest that species such as Paenibacillus brasilensis, Paenibacillus peoriae, and Paenibacillus kribbensis have potential for biocontrol of phytopathogens, as highly characterized BGCs, including Fusaricidin B, Bacillopaline, and Tridecaptin, were found with 100% similarity to previously described clusters.
Exploring the potential of Paenibacillus in the degradation of plant-based polymers
Carbohydrate-active enzymes (CAZymes) play a fundamental role in plant growth and defense, particularly in the agricultural context, where they contribute to the synthesis, remodeling, and degradation of structurally diverse carbohydrates. Through genome-wide analysis of complete Paenibacillus genomes, we identified 30 distinct CAZyme families (Supplementary Table S6), with glycoside hydrolases (GHs) emerging as the most dominant group, while polysaccharide lyases (PLs) were comparatively less represented.
Among all identified families, five were particularly abundant: GT2 (18.75%), GH43 (12.90%), GH32 (9.00%), GH13 (8.87%), and GH130 (7.50%), collectively accounting for 1,172 to 2,935 hits across the dataset (Fig. 5). These enzymes are functionally associated with the breakdown of a wide range of plant-derived polysaccharides, including cellulose, hemicellulose, arabinoxylan, inulin, sucrose, starch, glycogen, and mannose-rich oligosaccharides. Notably, species such as P. borealis, P. mucilaginosus, P. jilunlii, and P. polymyxa exhibited particularly high levels of these CAZyme families, suggesting a strong capacity for complex carbohydrate degradation (Fig. 5).Fig. 5. Bubble plot illustrating the abundance of genes involved in carbohydrate degradation across Paenibacillus strains. The abbreviation “GH” refers to glycoside hydrolases. The x-axis represents the genes, while the y-axis displays Paenibacillus strainsFig. 6. Heatmap illustrating the abundance of mobile genetic elements in Paenibacillus genomes (A). The figure correlates the number of plant growth promotion genes present in each element (Y-axis) and the isolation source of the strains (X-axis). The total quantity of each element identified in the strains (B)
In addition, the GH88 and GH95 families were especially enriched in these species, indicating their potential involvement in the degradation of uronic acid components of fucose-rich glycoproteins. These findings underscore the metabolic versatility of Paenibacillus and its potential relevance in biomass turnover and rhizosphere competence. Of particular interest was the widespread presence of GH18 family enzymes, which include bacterial chitinases—key players in biocontrol against fungal pathogens. Genes such as chiA, chiB, chiC, and chiD were detected in nearly all genomes, with P. guangzhouensis and P. mucilaginosus standing out due to their high content of GH18 enzymes, averaging 12 and 9 copies, respectively. These results strongly support the hypothesis that Paenibacillus chitinases contribute to both direct antagonism of phytopathogens and indirect plant growth promotion.
Mobile genetic elements in Paenibacillus genomes
Mobile genetic elements (MGEs) are major drivers of genomic plasticity, horizontal gene transfer, and microbial adaptation, making their analysis essential to understanding the dissemination of functional traits such as plant growth promotion and biocontrol potential in microbial populations. In this study, we investigated MGEs in Paenibacillus genomes to assess their diversity, genomic context, and potential contribution to ecological functions.
Using ICEscreen, we identified a total of 34 integrative elements, including nine complete integrative and conjugative elements (ICEs), seven partial ICEs, two complete integrative and mobilizable elements (IMEs), and sixteen degenerated or unclassified elements (Fig. 6). These elements ranged in size from 3,857 bp to 141,122 bp, with GC content varying from 36.0% to 54.4%. Although ICEs and IMEs exhibited limited representation of plant growth-promoting traits (PGPTs) and biocontrol-related genes, they frequently contained lplA and, in some cases, nod and pho genes (Supplementary Table S7), with a higher occurrence of genes in the categories competitive exclusion (23) and biofertilization (23), suggesting a potential, albeit modest, role in the dissemination of functional traits. None of the ICEs or IMEs showed matches in ICEBerg 3.0 and were therefore considered novel.
In parallel, twelve sequences related to transposable elements (transposons and insertion sequences) were identified, ranging from 353 bp to 1,770 bp and with GC content between 35.8% and 53.8%. These elements were predominantly recovered from soil and aquatic environments and showed no association with PGPTs or biocontrol genes.
In contrast, plasmids demonstrated greater functional relevance. A total of 35 plasmids were identified, showing remarkable size variation—from 973 bp to 2,697,188 bp—and GC content between 37.1% and 52.0%. These plasmids harbored a richer set of plant growth-promoting genes, comprising 44 genes distributed across specific categories such as nodulation and IAA production, and totaling 996 plant growth-promoting genes overall. Notably, one plasmid from Paenibacillus cellulosilyticus contained 137 bio-fertilization genes and 91 phytohormone-related genes (Supplementary Table S8). Furthermore, all plasmids included 15 biosynthetic gene clusters (BGCs) associated with the production of antimicrobial compounds such as paenilipoheptin, thermoactinoamide A, and bacillibactin.
Most functionally enriched plasmids were derived from strains isolated from soil (n = 33), feed ingredients (n = 13), and unknown sources (n = 9), underscoring the environmental relevance of MGEs in shaping the functional repertoire of P. populati. Additionally, four partial elements were found to be similar to each other and detected in different locations, although mostly recovered from the same type of environment (two from soil and two of unknown origin). This finding highlights the importance of MGEs as vectors for gene exchange, contributing to microbial community maintenance and ecological interactions within the genus Paenibacillus and its associated environments.
Pathogenicity and antibiotic resistance in Paenibacillus genomes
No virulence-associated genes were identified across the 97 Paenibacillus genomes analyzed. In contrast, a limited yet phylogenetically diverse repertoire of antibiotic resistance genes (ARGs) was detected. The most prevalent gene was rphB, associated with rifampicin resistance, present in 47 genomes (46.1%). This was followed by tetA and tetB, related to tetracycline resistance, detected in 14 genomes (13.7%). Additional ARGs were identified at lower frequencies and spanned resistance mechanisms to aminoglycosides (aadE-Pp), macrolides (mphI), lincosamides, phenicols, pleuromutilins (cipA, lsaB, TaeA), and glycopeptides (vanF/vanA) (Supplementary Table S9).
The strains with the highest number of ARGs were P. lautus, Paenibacillus sp. FSL W7-1332, and Paenibacillus sp. JZ16, each carrying up to 12 distinct resistance genes. These strains exhibited multidrug resistance profiles, including genes conferring resistance to tetracyclines, macrolides, aminoglycosides, lincosamides, and pleuromutilins.This finding suggests that, despite the generally low prevalence of ARGs across the genus, certain strains may serve as reservoirs of resistance genes. These results underscore the importance of monitoring gene mobility and potential environmental dissemination. For detailed annotations and gene distribution, refer to (Supplementary Table S9).
Discussion
Deciphering the biocontrol potential in Paenibacillus strains
Recent studies have demonstrated that Paenibacillus strains play a key role in the suppression of phytopathogens (Kim et al. 2019; Singh and Wesemael 2022a, b; Taheri et al. 2022a, b). Most of the current research has focused on P. polymyxa, a species that has attracted considerable attention due to its extensive potential across various industrial and agricultural sectors (Huang et al. 2024). However, there remains a clear need to investigate other species within the genus, particularly about their applications in sustainable agriculture.
To our knowledge, no systematic and comprehensive comparative genomic analysis has been conducted to investigate intrinsic features related to biocontrol and plant growth promotion across the Paenibacillus genus. In this study, we addressed this gap by selecting high-quality genomes to explore the functional potential of diverse Paenibacillus species. We first mapped the carbohydrate degradation profile of Paenibacillus. Our results revealed an extensive enzymatic repertoire among strains, with some encoding over 260 CAZymes, particularly in species also enriched in PGPT genes. The GH family (Glycoside Hydrolases), the largest and most studied group of CAZymes (Wardman et al. 2022), was the most dominant, with GH18 (chitinases) being ubiquitous across all strains. Chitinases hydrolyze β−1,4 glycosidic bonds in chitin and chitosan oligosaccharides, and play a key role in degrading fungal cell walls, insect exoskeletons, and nematode eggshells (Chen et al. 2020a, 2020b, 2020c; Moussian 2009).
The production of chitinases has been widely documented among Paenibacillus representatives (Du et al. 2021a, b; Veliz et al. 2017a, 2017b; Zhang et al. 2021). For example, P. elgii strain HOA73 was shown to control Gray Mold in tomato with efficacy comparable to that of commercial fungicides (Kim et al. 2019). Our comparative genomic analysis revealed a high abundance of GH18 enzymes in P. mucilaginosus, consistent with previous studies reporting its chitinolytic activity in vitro (Doan et al. 2019). The results also indicated that most Paenibacillus strains possess genes from GT2 and GH43 families, involved in the degradation of cellulose and hemicellulose, reinforcing the genus's capacity to utilize a broad range of carbon sources and adapt to soil environments.
Our analysis identified 128 distinct classes of secondary metabolites within the Paenibacillus genomes, reflecting the remarkable biosynthetic diversity of this genus. The most frequently detected clusters included NRPS, RiPPs, and polyketide hybrids, many of which are associated with antimicrobial activity. Several known bioactive compounds produced by Paenibacillus, such as fusaricidin, pelgipeptin, polymyxin B, tridecaptin, and colistin, were identified in our dataset (Dobrzyński and Naziębło 2024). Additionally, ribosomally synthesized peptides like paenicidin B were also observed (Lebedeva et al. 2021).
Secondary metabolites are low-molecular-weight bioactive compounds that, although not essential for primary metabolism, confer significant ecological advantages (Dewick 2009). The genes responsible for their biosynthesis are typically organized into discrete genomic regions known as biosynthetic gene clusters (BGCs) (Blin et al. 2023a, 2023b). Among these, non-ribosomal peptides (NRPs) are particularly abundant and include numerous antimicrobial agents and immunomodulators. NRPS (Non-Ribosomal Peptide Synthetases), modular enzymes responsible for the biosynthesis of these compounds, are widely recognized for their pharmaceutical and agricultural applications (Iacovelli et al. 2021).
Notably, some species stood out due to their high number of BGCs. P. mucilaginosus (18 BGCs), P. peoriae (17), P. polymyxa (17), and P. kribbensis (17) were particularly enriched. P. mucilaginosus, known for promoting plant growth, has been shown to produce NRPS-derived antimicrobials capable of suppressing Fusarium oxysporum (Wang et al. 2023b). Although the role of P. kribbensis BGCs in plant interactions remains unclear, many of its lassopeptides are known to inhibit microbial competitors (Mukhopadhyay et al. 2004). P. peoriae has been reported to possess antimicrobial activity against Pectobacterium brasiliense, attributed to its secondary metabolites (Zhao et al. 2022). In another study, P. peoriae HJ-2 showed antifungal efficacy against Fusarium concentricum in both greenhouse and field experiments involving Paris polyphylla, partly due to the presence of genes encoding fusaricidin synthetase (Jiang et al. 2022). Similarly, P. polymyxa WLY78 demonstrated that fusaricidin can induce systemic resistance via the salicylic acid (SA) signaling pathway against cucumber Fusarium wilt (Li and Chen 2019).
Finally, P. polymyxa remains the most widely studied species, with NRPS-based BGCs consistently linked to antimicrobial compound production, phytopathogen inhibition, and plant growth promotion (Jeong et al. 2019). These results emphasize the significance of Paenibacillus as a reservoir of biotechnologically relevant secondary metabolites and identify key species with promising applications in sustainable agriculture.
Main Paenibacillus species involved in plant interactions and their potential for agricultural applications
Most Paenibacillus species have been isolated from soil environments and are frequently found in the plant rhizosphere, suggesting that many members of this genus possess intrinsic capabilities for promoting plant growth. Our genomic analyses revealed a consistently high abundance of plant growth-promoting trait (PGPT) genes in various P. polymyxa strains, reinforcing the prominent status of this species. Widely acknowledged for its versatility and biotechnological relevance, P. polymyxa has been referred to as the "jack of all trades" by Langendries and Goormachtig (2021), owing to its multifaceted applications in both industry and agriculture.
Numerous studies corroborate the efficacy of P. polymyxa in promoting plant growth through a range of direct and indirect mechanisms. Direct contributions include the synthesis of phytohormones (Sun et al. 2022), efficient production of biofertilizers such as butanediol (Ju et al. 2023), exopolysaccharides (Liyaskina et al. 2021), and biological nitrogen fixation in crops such as cucumber (Li et al. 2022a, b). Additionally, the species can solubilize key nutrients like phosphorus and zinc (Ahmad et al. 2021).
Species such as P. mucilaginosus, P. thiaminolyticus, and P. borealis exhibited the largest sets of genes associated with plant growth-promoting traits, particularly those related to biofertilization, indicating a potentially more relevant role in supporting plant growth. Although P. polymyxa remains the most studied and widely applied species, our results highlight that other species, such as P. peoriae and P. kribbensis, display comparable functional potential — each harboring the same total number of PGPT-related genes as P. polymyxa. Both P. peoriae and P. kribbensis have been investigated for their biocontrol properties (Yurong et al. 2022), and P. kribbensis is noted for its capacity to produce a wide array of secondary metabolites with antibacterial activity (Li et al. 2024). However, their roles as plant growth-promoting rhizobacteria (PGPRs) remain largely unexplored.
Additional species—such as P. jilunlii, P. sophorae, and P. brasilensis—also exhibited elevated frequencies of PGPT-related genes. Notably, P. brasilensis is a nitrogen-fixing bacterium associated with maize (Von der Weid et al. 2002) and has been shown to delay senescence in tangerines (Chen et al. 2020a, b, c), emphasizing the functional diversity within the genus. On average, the genomes analyzed in this study encoded approximately 96 PGPT-related genes, suggesting that the capacity for plant growth promotion is broadly distributed across Paenibacillus species.
Emerging evidence from the literature further supports the agricultural potential of less-explored species. For instance, Paenibacillus monticola enhances white clover seedling growth (Li et al. 2022a, 2022b), P. lentimorbus mitigates nutrient deficiencies in maize (Singh et al. 2024), and P. mucilaginosus, when co-inoculated with other beneficial rhizobacteria, improves soybean agronomic performance (Xing et al. 2022). As emphasized by Dobrzyński and Naziębło (2024), there is a pressing need to expand the scope of research beyond P. polymyxa to uncover the hidden potential of other Paenibacillus species. Our findings support this view, indicating that a broader array of species within the genus may be effectively employed in both the biocontrol of phytopathogens and the direct promotion of plant growth in agricultural systems.
Phosphate solubilization and iron acquisition: predominant mechanisms of Paenibacillus in plant growth promotion
Phosphorus and iron are essential micronutrients required for optimal plant development (Sharma et al. 2013; Sultana et al. 2021). Despite their abundance in the soil, these elements are often inaccessible to plants due to their chemical forms. Phosphorus is typically immobilized through complexation with metal ions or incorporated into organic matter, rendering it insoluble (Rawat et al. 2021). Similarly, under alkaline conditions, iron (Fe^3^⁺) tends to precipitate as insoluble oxyhydroxides (Grady et al. 2016). This limited bioavailability frequently necessitates the application of agrochemicals to supplement phosphate or to correct soil pH (Sharma et al. 2013).
In this context, the use of bioinoculants has emerged as a sustainable and cost-effective alternative for enhancing nutrient availability in agricultural systems (Elnahal et al. 2022; Singh and Kumar 2024). Numerous Paenibacillus species have been reported for their capacity to solubilize inorganic phosphate and facilitate iron uptake (Grady et al. 2016; Soni et al. 2021; Yuan et al. 2022; Wang et al. 2023a, b; Wendisch et al. 2023; Dobrzyński and Naziębło 2024). Phosphate solubilization in Paenibacillus is often attributed to the production of organic acids such as gluconic acid, while iron acquisition may involve siderophore secretion, organic acid exudation, or induction of plant genes related to iron mobilization (Grady et al. 2016).
Our genomic analyses indicate that phosphate solubilization in Paenibacillus is predominantly mediated by organic acid production, mainly through the PQQ-dependent glucose oxidation pathway, which leads to gluconic acid synthesis and medium acidification. Genes associated with this pathway, including gcd and multiple pqq genes (pqqC, pqqD, pqqE, and pqqL), as well as genes involved in gluconate and keto-gluconate metabolism (gnl, gnd, gadh, and ghrB), were detected at high frequency across the analyzed genomes, supporting a conserved mechanism for inorganic phosphate solubilization. In addition, genes encoding phosphatases and genes involved in polyphosphate metabolism (e.g., ppk and ppx) were identified with a more heterogeneous distribution, suggesting complementary and species-specific phosphate acquisition strategies within the genus. Consistent with these mechanistic insights, our analyses indicate that the genetic potential for phosphate solubilization and iron acquisition is widely distributed across the genus. Notably, P. polymyxa, P. kribbensis, P. brasilensis, and P. graminis harbour an extensive repertoire of genes associated with phosphate solubilization and iron acquisition. Among these, the genes phy, phyA, phyB, and phyC were particularly abundant and recurrent. Experimental validation has confirmed phosphate solubilization in P. polymyxa (Mohd Din et al. 2020), P. kribbensis (Zhang et al. 2013), and P. brasilensis (Arthurson et al. 2011). In contrast, iron acquisition has been extensively characterized only in P. polymyxa (Zhou et al. 2016; Grady et al. 2016), indicating a knowledge gap regarding the expression of this trait in other Paenibacillus species.
The potential of Paenibacillus spp. for plant growth promotion, particularly through these nutrient acquisition strategies, is well documented (Grady et al. 2016). For example, P. polymyxa has demonstrated beneficial effects in several agronomically important crops, including rice (Abdallah et al. 2019), maize (Mohd Din et al. 2020), pepper (Phi et al. 2010a, 2010b), tobacco (Liu et al. 2020), and wheat (Li et al. 2023). However, despite the genomic potential observed in P. kribbensis, P. brasilensis, and P. graminis, their functional capacities remain underexplored.
Further in vitro and in vivo studies are necessary to assess the extent to which these lesser-studied species can solubilize phosphate and mobilize iron under field-relevant conditions. Such efforts could lead to the identification of novel, effective candidates for the development of next generation biofertilizers, contributing to more sustainable agricultural practices.
Key characteristics that make Paenibacillus a promising genus for biotechnological applications
Species of the genus Paenibacillus have garnered growing interest over the past decades, primarily due to their biotechnological potential (Bloemberg and Lugtenberg 2001). Several intrinsic features support their candidacy as versatile tools for agricultural and industrial applications. One such characteristic is their motility via flagella, which enables Paenibacillus strains to detect and respond to root exudates and mucilage components, facilitating chemotactic movement and efficient root colonization. Another crucial trait is their ability to form endospores, which enhances their resilience to environmental stresses and improves their viability and shelf life in bioformulations (Dobrzyński and Naziębło 2024). This spore-forming capacity makes them highly suitable for large-scale applications in diverse and fluctuating field conditions.
In addition to traits directly associated with plant growth promotion, functional annotation based on the PGPG_finder pipeline revealed a consistent set of indirect effects related to environmental adaptation. Genes associated with root system colonization and the utilization of plant-derived substrates, such as araR and the lplA–C gene cluster, were widely distributed among the 97 analyzed genomes, suggesting an adaptive capacity related to the rhizosphere. This pattern is reinforced by the heatmap shown in Supplementary Fig. 1, which provides an integrated overview of direct and indirect functional effects across strains and highlights the high abundance of categories related to abiotic stress mitigation, motility, and chemotaxis. Consistently, genes involved in chemotaxis and motility (tlpA, tlpC, and drcA), stress response (rhlE and nht), and salinity and halotolerance (yfbK, atpC, and atpD) were detected in all genomes or in at least one copy per genome. The broadly conserved distribution of these genes, detailed in Supplementary Table S3, reinforces the genomic plasticity and ecological versatility of Paenibacillus species.
Mobile genetic elements (MGEs), including plasmids, ICEs, IMEs, and transposons, are key mediators of horizontal gene transfer (HGT) and can facilitate the dissemination of adaptive traits among bacterial populations (Dobrindt et al. 2004; Simpson et al. 2025). In this study, we observed that several genes associated with plant growth promotion and biocontrol were located on MGEs, particularly plasmids, which harbored a diverse set of genes involved in nodulation, IAA production, and other plant growth-promoting functions (Rodríguez-Beltrán et al. 2021; MacLean and San Millan 2019). The presence of these genes on MGEs highlights the potential of horizontal transfer to spread beneficial traits across Paenibacillus populations, reinforced by the detection of the same element in isolates from four different locations.
This genetic mobility may enhance the ecological versatility of strains, enabling adaptation to diverse environments and the establishment of important ecological interactions (Arnold et al. 2021). Furthermore, the use of strains containing MGEs as bioinoculants offers an additional advantage, as these bacteria could transfer beneficial traits to the resident soil microbiome without the need for reapplication, or even by utilizing only the MGEs from these strains (Brophy et al. 2018). Overall, our findings underscore the importance of considering MGE-associated genes when evaluating the ecological potential and biotechnological applicability of Paenibacillus strains.
The open configuration of the Paenibacillus pan-genome, supported by our findings, underscores the genus's genomic plasticity and its ongoing capacity to acquire novel genes from its environment. This genomic flexibility is vital for adaptation to the heterogeneous and competitive conditions of soil and rhizosphere ecosystems. A significant portion of both the core and accessory genomes is dedicated to functions associated with carbohydrate transport and metabolism (COG category G), reflecting an ecological strategy centered on nutrient acquisition and environmental sensing (Park and Kong 2018). Additionally, the remarkable biosynthetic potential for producing a wide array of bioactive secondary metabolites further enhances the genus's appeal for biotechnological innovation. These metabolites include antimicrobial compounds, siderophores, and phytohormones, which collectively support Paenibacillus in promoting plant growth, suppressing pathogens, and modulating plant immune responses.
Conclusion
Our comprehensive comparative genomic study highlights Paenibacillus as a key microbial genus in plant–microbe interactions, with robust capabilities for promoting plant growth and suppressing phytopathogens through multifaceted mechanisms. The data presented herein emphasize the ecological relevance, genomic diversity, and biotechnological potential of this genus. These insights contribute to a deeper understanding of Paenibacillus biology and set the foundation for its application in sustainable agriculture. Future research should focus on isolating and characterizing novel Paenibacillus species, optimizing bioinoculant formulations, and evaluating their efficacy under field conditions. Furthermore, the impact of Paenibacillus-based products on native soil microbiota, ecosystem dynamics, and animal and human health warrants investigation. Altogether, this study supports the integration of Paenibacillus spp. into next-generation agricultural strategies aimed at reducing chemical inputs and enhancing crop productivity in a sustainable manner.
Supplementary Information
Below is the link to the electronic supplementary material.Supplementary file1 (PDF 33 KB)Supplementary file2 (PDF 1798 KB)Supplementary file3 (XLSX 1925 KB)