The insider's perspective: The intracellular complosome and immune cell dynamics in cancer
Alexandra Bennion, Joanne Lysaght, Niamh Lynam‐Lennon

TL;DR
This paper explores how intracellular complement (complosome) influences cancer immunity, affecting immune cell function and tumor growth.
Contribution
The paper introduces the complosome as a novel target for cancer immunotherapy by detailing its role in immune cell dynamics.
Findings
Complosome activity rewires T-cell metabolism and promotes pro-tumor immune cell function.
Blocking the complosome reveals a new therapeutic target in cancer.
C3/C5-driven complosome signals influence T-cell activation and tumor immunity.
Abstract
Complement is increasingly recognised as a driver and modulator of antitumour immunity, with context‐dependent effects across T cells, myeloid subsets, stromal elements and tumour cells. Although best known for pathogen clearance and membrane attack complex (MAC) formation, complement also acts intracellularly via the ‘complosome’ to regulate cellular homeostasis and gene expression. Complosome activity may dampen antitumour responses by rewiring single‐cell metabolism and transcription, altering nutrient flux and fostering an immunosuppressive microenvironment. Here, we synthesise advances in intracellular and extracellular complement, with emphasis on complement component 3 (C3) and receptors (C3aR1, C5aR1/CD88, C5aR2/C5L2), highlighting how these pathways shape T‐cell metabolism, exhaustion programmes and inflammatory tone within tumours. Evidence indicates that tonic C3/C5…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
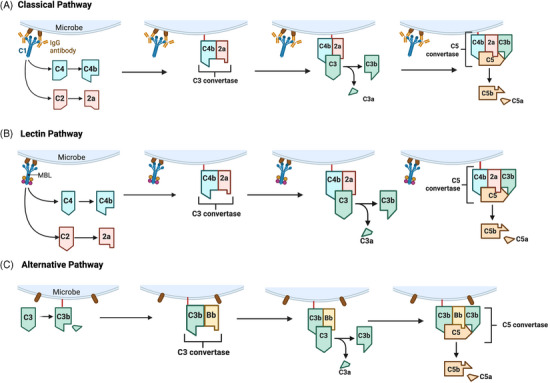 FIGURE 1
FIGURE 1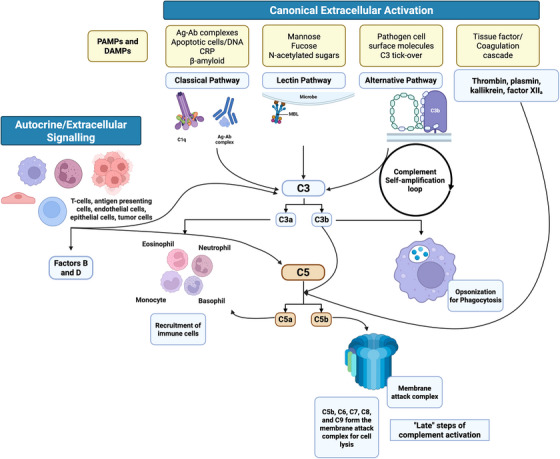 FIGURE 2
FIGURE 2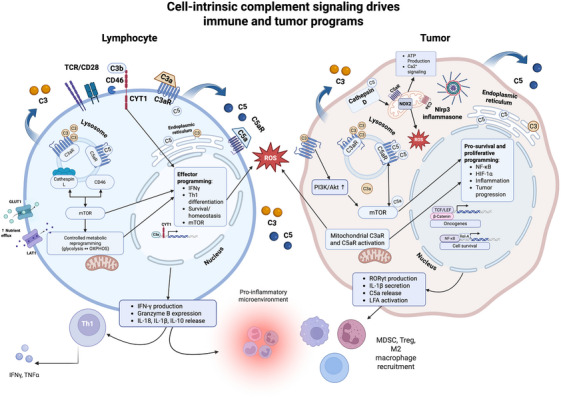 FIGURE 3
FIGURE 3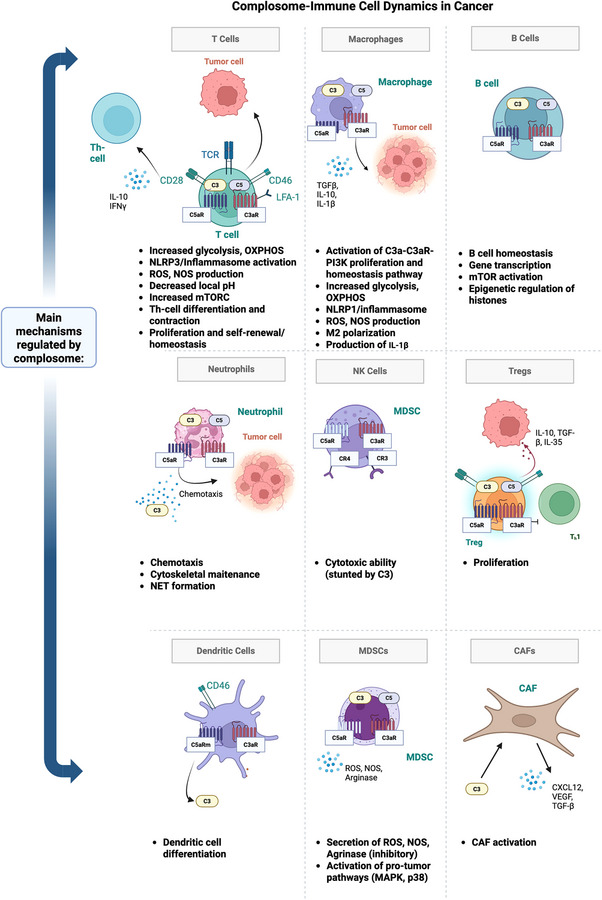 FIGURE 4
FIGURE 4| Protein | Cancer type | Description | References |
|---|---|---|---|
|
| |||
| C1q | Cervical cancer | Increased levels of C1qB protein are found in serum and cervical cancer tumour tissue. C1qB expression positively correlated with Ki67 and P16 expression |
|
| Melanoma, colon adenocarcinoma, lung adenocarcinoma, breast adenocarcinoma, pancreatic cancer | Increased levels of C1q in tumour stroma and vascular endothelium, mainly expressed by vascular endothelial cells, fibroblasts and myeloid cells |
| |
| Glioblastoma | Serum C1q upregulated in glioblastoma patients vs. healthy matched tissue, and C1q marked deposition within tumour tissue |
| |
| C1s | Cutaneous squamous cell carcinoma (cSCC) | Overexpression of C1s in cSCC observed in cell lines and patient samples. Knockdown of C1s promotes apoptosis and growth suppression in vitro |
|
| Renal cancer | Overexpression of C1s in tumours independently associated with high infiltration of macrophages and T cells in patient samples and associated with poor prognosis |
| |
| Prostate cancer | Serum C1q upregulated in prostate patients vs. healthy matched tissue |
| |
| C1qBP | Lung cancer | Overexpression of C1qBP in patient lung cancer samples compared to healthy tissue |
|
| C1r | SCC | Overexpression of C1r in SCC observed in cell lines and patient samples. Knockdown of C1r promotes apoptosis and growth suppression in vitro |
|
| Melanoma | Overexpression of C1r in patient samples of melanoma vs. healthy tissue | ||
| C3 | Gastric cancer | Overexpression of C3 in gastric cancer tissue compared to healthy tissues |
|
| Pancreatic cancer | C3 overexpressed in PDAC tissue, and its expression correlates with metastatic potential |
| |
| C3a | cSCC | C3a overexpression in patient tumour tissues correlated with tumour cell growth in vitro |
|
| Oesophageal adenocarcinoma | Overexpression of C3a in oesophageal adenocarcinoma tissue compared to healthy tissues |
| |
| Breast cancer | Overexpression of C3a in serum of breast cancer patients compared to healthy patients |
| |
| C3b | NSCLC | Overexpression of C3 observed in paclitaxel resistance of NSCLC cells; knockdown of C3 promotes apoptosis |
|
| C4a | Papillary thyroid cancer | Overexpression of C4a in serum of patients |
|
| C4d | Renal cancer | Overexpression of C4d in plasma of renal cancer patients and deposits at tumour site |
|
| C9 | Gastric cancer | C9 overexpression in the sera of gastric cancer patients vs. healthy controls |
|
| CD55 | Colon cancer | CD55 overexpression associated with less differentiated, higher grade tumour tissue in patient samples |
|
| CD59 | Colon cancer | CD59 overexpression associated with less differentiated, higher grade tumour tissue in patient samples |
|
| CD46 | Bladder cancer | CD46 overexpressed in bladder cancer compared to healthy tissue, and CD46 expression inversely correlated with tumour stage, grade and disease progression risk |
|
|
| |||
| C1q | Prostate cancer | C1q downregulated in benign prostatic hyperplasia and prostate cancer tissue, inactivating tumour suppressor WWOX |
|
| C1s | Ovarian cancer | Downregulation of C1s mRNA observed in diseased tissue vs. healthy control |
|
| Lung cancer | Downregulation of C1s at site of lung cancer tumour tissue compared to peritumoral tissue |
| |
| C4BP | Ovarian cancer | Downregulation of C4BP mRNA in ovarian cancer tissue compared to healthy control |
|
| C7 | Ovarian cancer | Downregulation of C7 mRNA in ovarian cancer tissue compared to healthy control |
|
| CD55 | Ovarian cancer | Downregulation of CD55 in ovarian cancer tissue compared to healthy controls |
|
- —U.S.–Ireland Alliance
- —Health Research Board10.13039/100010414
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComplement system in diseases · Monoclonal and Polyclonal Antibodies Research · Malaria Research and Control
INTRODUCTION TO THE COMPLEMENT SYSTEM
1
One of the oldest and most important components of innate immunity is the activation of the complement system, a cluster of over 50 highly conserved, membrane‐bound and intracellular proteins that primarily circulate in blood and lymph, opsonise pathogens and induce inflammatory responses.1 Initially described as a ‘complement’ to enhance and support antibodies in the detection and removal of pathogens, it is now understood that the complement system can independently trigger immunosurveillance.
Three known canonical pathways activate liver‐derived and serum‐effective complement in the extracellular space: (i) the classical (antibody‐dependent) pathway, (ii) the lectin (carbohydrate recognition) pathway, and (iii) the alternative pathway activated continuously at low levels2 (Figure 1).
Overview of the three main complement activation pathways including the (A) classical pathway, (B) lectin pathway and (C) alternative pathway. All three pathways converge at the cleavage of C3. Unlike the classical or lectin pathway, the alternative pathway is continuously active at low levels due to spontaneous hydrolysis of C3 thioester and utilises a distinct C3 convertase comprised of C3b and a cleaved fragment of plasma protein Factor B. When C3b is bound to a pathogen surface, Factor B binds and is cleaved by plasma protein Factor D, resulting in C3bBb (C3 convertase). Complement may also be activated via the coagulation cascade pathway (tissue damage) Factor XIIa, which upregulates C1 in the classical pathway, or via inducible serine proteases and proteins (thrombin, plasmin) activated by macrophages. 148 Created with BioRender.com.
All pathways converge on the central component C3, generating activation products C3a/C3b and downstream C5a/C5b, culminating in anaphylatoxin release, opsonisation and membrane attack complex (MAC) assembly. Beyond hepatic sources, extracellular complement proteins (namely, C3 and C5) can also be secreted by macrophages, monocytes, endothelial cells, epithelial cells, natural killer (NK), dendritic cells (DC), cancer cells, B and T lymphocytes3, 4 (Figure 2).
An overview of extracellular complement activation. Canonical complement activation via the classical, lectin and alternative pathways and the coagulation cascade. Autocrine complement protein secretion occurs in T cells, antigen‐presenting cells and endothelial cells. Created with BioRender.com.
INTRACELLULAR COMPLEMENT: THE COMPLOSOME
2
While historical attention has focused on the complement system as an extracellular component of innate immunity, intracellular complement known as ‘the complosome’ has been recently identified as a key cell‐intrinsic signalling programme that integrates metabolism, autophagy, immune activation and gene regulation.1 First defined in CD4^+^ T cells in 2013, intracellular complement activation and endogenous C3 and C5 protein production were revealed as essential for T‐cell homeostasis and pro‐inflammatory cytokine production.5 Since then, complosome activity has been detected in monocytes, macrophages, dendritic cells, epithelial and cancer cells, among others.1, 6
Generation of intracellular complement effectors
2.1
Intracellular complement signalling is mediated by C3aR1 and C5aR1/C5aR2, which can signal from endosomal/lysosomal compartments and, in defined contexts, from mitochondria or the plasma membrane of immune and non‐immune cells.7 The transmembrane cofactor/receptor CD46 is a second hub of complosome control: C3b (and C4b) engagement of CD46 triggers intracellular cascades (via CYT‐1/CYT‐2 cytoplasmic tails) that couple nutrient sensing and transcriptional programmes to complement status.8 T‐cell receptor (TCR) stimulation in T cells and Toll‐like receptor (TLR) activation in antigen‐presenting cells can drive de novo C3/C5 generation, with cathepsin‐dependent cleavage proposed as one route to yield C3a/C3b/C5a intracellularly; precise enzymology remains incompletely defined.9 In parallel, stores of C3 and C5 have been observed within intracellular vesicles (endosomes and lysosomes) of multiple cell types, facilitating intracellular complement activation when transported out of the cell membrane or vesicle, where autocrine re‐engagement with C3aR and C5aR is possible.5 CD46 signalling (via CYT‐1 nuclear shuttling and metabolic cues) has been linked to upregulated C3/C5 transcription, further reinforcing the complosome loop.10
COMPLEMENT AND CANCER
3
Mounting evidence places the intracellular complement programme (complosome) at the interface of tumour biology and immunity, linking cell‐intrinsic signalling to T‐cell dysfunction, myeloid remodelling, and therapy resistance in the tumour microenvironment. Local complement activity is detected across cancers, supporting a role in the tumour microenvironment (TME) (Table 1).
In the clinic, among glioblastoma multiforme patients, elevated C3 and C5b levels were observed in tumour tissue versus healthy controls.13 Elevated deposition of C3 has also been observed in patient samples of non‐small cell lung cancer (NSCLC), gastric cancer and pancreatic cancer.18, 19, 34 High deposition of C3 has been significantly associated with poor overall 5‐year survival in gastric cancer patient samples, and was significantly associated with pancreatic cancer metastasis.18, 19 The effector anaphylatoxin C3a has also been shown to have increased deposition in tumour cell tissue when compared to healthy control tissue in cutaneous squamous cell carcinoma (cSCC), breast cancer and oesophageal adenocarcinoma.35 Additionally, elevated complement proteins are frequently found in sera from cancer patients versus healthy controls. Elevated C3a protein has been observed in the sera of breast cancer patients, and elevated C3a in breast cancer tissue is associated with lymph node metastasis.23, 36
Functionally, complement proteins produced both intracellulary and extracellularly have been shown to have a direct impact on tumour growth and therapeutic response. In a lung cancer murine model, silencing of C3 inhibited tumour growth,37 and in NSCLC models, nuclear C3b is enriched in paclitaxel‐resistant cells, implicating complement in drug resistance.24 Clinically, complement proteins have been linked to treatment response and resistance. In colorectal (CRC) cancer, high C3 expression was associated with increased FOLFOX chemotherapy resistance and poorer prognosis.38 Although complement classically enables cell killing, the complosome in immune and tumour cells can promote a pro‐tumour, immunosuppressive TME via myeloid recruitment, T‐cell metabolic/exhaustion programmes and altered cytokine networks.39 Here, we map the complosome–immune microenvironment interface in cancer, examining how these circuits may underpin treatment resistance, tumour response and inform actionable interventions.
Despite increasing evidence supporting a pro‐tumour role for intracellular complement signalling, it is important to highlight that findings across studies are not uniformly concordant. While multiple reports demonstrate that intracellular C3 and C5 activation promotes tumour cell survival, immune evasion and metabolic adaptation, other studies suggest that complement signalling may exert context‐dependent or even antitumour effects under specific conditions.40, 41 For example, discrepancies have been reported depending on tumour type, stage and immune composition, as well as whether complement activity is assessed acutely versus chronically.42, 43 These divergent observations highlight that complosome signalling is not inherently oncogenic, but instead reflects a context‐sensitive regulatory axis whose functional consequences depend on cellular state and microenvironmental cues.
COMPLEMENT, IMMUNE ACTIVATION AND T CELLS IN CANCER
4
Rather than functioning through isolated signalling pathways, intracellular complement activation integrates metabolic, transcriptional and inflammatory programmes that collectively shape immune cell fate and tumour behaviour. In both immune and malignant cells, the complosome acts as a central organising axis that links intracellular proteolysis of complement components to downstream metabolic rewiring, cytokine output and cell survival. Understanding how these pathways converge, rather than considering them as independent observations, is essential for appreciating the role of the complosome in immune activation, immune suppression and therapy resistance.
Immunologically, intracellular complosome and complement proteins secreted by tumour and/or immune cells play a significant role in the housekeeping of immune responses, particularly among T cells. CD4^+^ and CD8^+^ T cells are the two lymphocyte subsets that are crucial for cell‐mediated immunity and the activation of cytotoxic immune responses to tumour cells.44 CD4^+^ and CD8^+^ T cells contain low but measurable intracellular stores of C3/C5, with de novo synthesis upregulated following TCR or cytokine stimulation.3, 45 It has been shown that intracellular cathepsins (CTSL) can then cleave C3/C5 to generate C3a/C5a, supporting cell‐intrinsic complosome signalling.5, 46
Early infection models implicated complement in T‐cell responses; C3 was first suggested to mediate CD4^+^ and CD8^+^ T‐cell responses in 2002, where mice deficient in C3 had significantly lower lymphocyte responses in a preclinical model of influenza.47 However, the link between complement and T‐cell‐mediated anticancer immunity was not identified until 2008 by Markiewski et al., when TME‐derived C5a was shown to suppress antitumour CD8^+^ T‐cell responses, enhance tumour growth, increase production of reactive oxygen species (ROS), nitric oxide synthase (NOS) and immunosuppressive myeloid‐derived suppressor cells (MDSCs) in a murine cervical cancer model.48 Subsequent work has linked complement to dampened anticancer CD4^+^/CD8^+^ responses across models, including C5aR1‐dependent suppression and C3a‐driven restraint of CTL proliferation/apoptosis programmes.48, 49, 50 Vadrevu et al. demonstrated that expression of C5aR promotes metastases in a breast murine model by suppressing CD8^+^ and CD4^+^ T‐cell populations and function in lung tissue.49 C3a has also been shown to be specifically expressed by immune cell populations and promotes T‐cell apoptosis and inhibition of CTL proliferation.51
Potential complosome–immune axis mechanisms underlying carcinogenesis
4.1
Defects in antitumour CD8^+^/CD4^+^ function reflect complosome‐connected mechanisms: (i) T‐helper differentiation, (ii) metabolic reprogramming, (iii) APC‐mediated suppression and (iv) CD46 signalling52 (Figure 3). In T cells, intracellular complosome activity is mainly mediated by C3aR1/C5aR1/C5aR2, which can signal from the plasma membrane, endosomes/lysosomes and in defined contexts, mitochondria.5 Upon activation of these receptors on and within T cells, increases in cellular glycolysis, oxidative phosphorylation (OXPHOS), mTOR signalling, nutrient efflux, ROS and pro‐inflammatory cytokines (context‐dependent [IL‐12, IL‐13]) have been observed.53 However, T cells are not the only immune subpopulation shown to mediate or be mediated by complosome activation. In a sentinel study by Liszeski et al., both basal ‘resting state’ intracellular stores of C3 and ‘tonic’ intracellular C3a have been observed in myeloid (monocytes, neutrophils), lymphoid (CD8^+^ T cells, B cells), epithelial, endothelial cells and fibroblasts.5 While the complosome is most well characterised in T cells, intracellular complement seems to be a ubiquitous phenomenon across immune cell populations. Additionally, complosome activity within tumour cells themselves has the potential to recruit immunosuppressive MDSCs,54 impair T‐cell function55 and polarise macrophages to a pro‐tumour phenotype.56
An overview of the complosome signalling dynamics governing immune activation in both immune and cancer cells. Schematic illustrating parallel but functionally divergent complosome signalling pathways in lymphocytes (left) and tumour cells (right). In lymphocytes, intracellular stores of C3 and C5 are cleaved within endolysosomal compartments, generating C3a and C5a that signal through intracellular and surface‐localised C3aR and C5aR, as well as CD46. Engagement of these receptors coordinates metabolic reprogramming, including increased glycolysis, nutrient uptake via GLUT1 and LAT1, mTOR activation, and controlled reactive oxygen species (ROS) production, supporting T‐cell survival, Th1 differentiation and effector functions such as IFN‐γ production and cytotoxic mediator release. In tumour cells, cell‐intrinsic complement activation similarly engages C3aR and C5aR localised to endosomes, lysosomes, mitochondria and the endoplasmic reticulum. Downstream intracellular complosome signalling preferentially activates pro‐survival and pro‐tumorigenic pathways, including PI3K‐AKT‐mTOR signalling, NF‐κB and HIF‐1α transcriptional programmes, inflammasome activation and sustained ROS production. These pathways promote metabolic adaptation, resistance to stress, inflammatory signalling and tumour progression, while shaping an immunosuppressive microenvironment through recruitment of myeloid‐derived suppressor cells (MDSCs), regulatory T cells and M2‐polarised macrophages. Key unanswered questions include the temporal regulation of intracellular complement activation and the extent to which complosome signalling is conserved across tumour types and immune subsets. Created with BioRender.com.
Collectively, these findings support a model in which intracellular C3 and C5 cleavage initiates a cascade of receptor‐dependent signalling events that converge on metabolic reprogramming and transcriptional control. In lymphocytes, this integration canonically supports controlled effector differentiation and subsequent resolution, which may be dysregulated in cancer contexts, whereas in tumour cells, chronic complosome activation reinforces inflammatory signalling, metabolic autonomy and immune suppression.9, 57 Framing these observations within a unified mechanistic model clarifies how individual molecular findings relate to broader immune and tumour phenotypes.
Complosome, homeostasis and Th differentiation
4.2
Intracellular complement activation is an important mediator of T‐cell differentiation (particularly antitumour Th1 production, a subset of CD4^+^ T cells) and maintenance, tightly regulated by the production of interferon (IFN)‐γ and intracellular C3 cleavage.58, 59
Upon TCR and CTSL (ubiquitously expressed lysosomal endopeptidase) continuously cleaves intracellular stores of C3 into effector C3a and C3b, which are either exported to the cell surface or initiate intracellular transcription of interferon IFN‐γ following Th1 differentiation.5 As such, C3‐deficient systems show impaired Th1 and reduced IL‐4/IL‐5/IL‐13/IFN‐γ after antigen stimulation. For example, C3‐knockout mice have been shown to secrete less IL‐4, IL‐5, IL‐13 and IFN‐γ in response to antigen stimulation, limiting potential antitumour Th1 response dependent on IFN‐γ.60 In people, patients deficient in C3 or CD46 (co‐stimulator of IFN‐γ Th1 cell induction) have been shown to be deficient in Th1 cells and experience frequent infections.61, 62 Inhibition of C3 in vivo has also been shown to reduce Th cells producing IL‐4 (Th2), IL‐17 (Th17), IL‐2 and TNF‐α, reduce naïve CD4^+^ cells, and reduce the proliferation of both CD4^+^ and CD8^+^ cells.63 Along with intracellular C3, C3aR and C5aR1 have also been shown to be required for adequate Th1 differentiation. Mice deficient in either C3aR or C5aR1 on T cells and APCs exhibited reduced Th1 expansion in multiple in vivo studies.64, 65 In a model of bacterial response to Listeria monocytogenes (LM), C3‐deficient mice showed blunted Ag‐specific CD4^+^/CD8^+^ expansion during infection.66
In the context of cancer, elevated Th1 cells have been associated with a favourable prognosis in multiple cancer types, including NSCLC, ovarian cancer, breast cancer, melanoma, glioblastoma and CRC.67, 68, 69, 70 Thus, dysfunctional Th differentiation, mediated by dysfunctional intracellular complement signalling or C3 protein deficiency, is a plausible mechanism of immunosuppression in cancer.
Aside from differentiation, intracellular complement is an important component of homeostasis. Intracellular C3a has been shown to engage with C3aR on lysosomes to activate and sustain mTOR, a potent regulator of glycolysis, inhibitor of immunosuppressive Treg differentiation and promoter of T‐cell survival and homeostasis.5, 71 When C3a is blocked, swift T‐cell apoptosis occurs.5 Importantly, extracellularly produced C3a from serum is unable to rescue and reinstate T‐cell proliferation in T cells deficient in intracellular C3/C3a,5 indicating that the intracellular C3/C3a axis is necessary to maintain T‐cell function.
Complosome and metabolic reprogramming of T cells
4.3
T‐cell fate and effector functions of all immune cells are coupled to cellular metabolism, and there is growing evidence that shifts in metabolic pathways by the complosome shape adaptive immune responses of CD8^+^ cells.72 In the inactive state, T cells preferentially rely on OXPHOS to generate ATP,73 whereas activated and proliferating T cells are known to upregulate glycolysis.73, 74 Chang et al. demonstrated that when glycolysis is blocked in vitro by galactose, CD4^+^ T cells exhibit minimal IFN‐γ and IL‐2 production, stunting Th1 expansion, and CD8^+^ cells show reduced proliferation, showcasing the increased glycolytic need to sustain effector function.75
There is growing evidence that the complosome may be implicated in this metabolic shift towards glycolysis, facilitated by intracellular stores of C3/C3a in CD4^+^ and CD8^+^ T cells (predominantly produced by lysosomes and the endoplasmic reticulum).9 Complosome signalling contributes to this metabolic shift, as C3aR1/C3 engagement (in T‐cell lysosomal compartments) has been shown to maintain basal mTOR (regulator of glycolysis) and GLUT1 (glucose transporter 1) expression.46 However, it is unknown if upregulation of C3aR or intracellular C3 drives aberrant hyperactivation of glycolysis. TCR activation results in the cleavage of stored intracellular C5 by cathepsin D, generating C5a. C5a binds to mitochondrial C5aR1 and has been shown to activate glycolytic flux in neutrophils, and it is likely this process also occurs in T cells due to similar C5aR1 expression levels; however, this has still to be demonstrated in the T‐cell population.76, 77
Stimulation of TCR and co‐stimulation of CD28 in CD4^+^ T cells shuttles intracellular C3b (produced from endogenous C3) to cell‐surface CD46.78 CD46 couples complement to nutrient programmes: C3b–CD46 triggers γ‐secretase cleavage and CYT‐1 nuclear translocation, inducing LAT1, GLUT1 and LAMTOR1, thereby increasing amino acid/glucose uptake and supporting Th1 differentiation and IFN‐γ.1 In a study by Kolev et al., activation of CYT‐1 was required for the activation of LAT1 and increased expression of GLUT1 and LAMTOR1 in human T cells.46 When GLUT1 and LAT1 are upregulated, nutrient efflux enhances intracellular uptake of glucose and essential amino acids to support increased glycolysis and Th1 differentiation.79 Unsurprisingly, T cells from patients with CD46 deficiency show dysfunctional Th1 cell induction and decreased glycolysis and OXPHOS.80
Maintenance of T‐cell homeostasis by the complosome introduces a delicate balance of pro‐ and antitumorigenic signals. While Th1 expansion is associated with antitumour responses, the upregulation of GLUT1 and LAMTOR1 is associated with the induction of an immunosuppressive microenvironment and hyperproliferation in both immune and tumour cells.81
C3aR/C5aR axis and immunosuppression
4.4
C3aR and C5aR are highly expressed on tumour and immune cells in preclinical and patient samples.82, 83, 84 Interestingly, while preclinical studies suggest C3aR and C5aR are significantly upregulated in murine and patient tumours and tumour‐infiltrating lymphocytes (TILs), peripheral CD8^+^ T cells from non‐tumour‐bearing mice exhibit a lack of expression of both receptors.85 Activation of C5aR intracellularly by C5 cleavage by cathepsin D (CTSD) has been shown to promote carcinogenesis in CRC, via the formation of a C5a/C5aR1/KCTD5/cullin3/Roc‐1 complex, which stabilises β‐catenin and drives downstream oncogenic signalling.76 The increased transcription of stabilised β‐catenin results in enhanced transcription of target oncogenes Cox2, cyclin D1 and c‐Myc.76 Intrinsic WNT/β‐catenin signalling in tumour cells is also associated with poor T‐cell infiltration, and WNT/β‐catenin signalling in T cells has been shown to promote the expression of RORγt, a signature of Th17 cells (potent activators of Tregs).86, 87
Inflammasome crosstalk adds complexity to the immunosuppressive function of the complosome. The C5aR signalling axis is required for the assembly of the intracellular protein ‘inflammasome’ complex, which promotes maturation of pro‐inflammatory IL‐18, IL‐1β, release of pro‐inflammatory HMGB1 protein and caspase‐1‐dependent pyroptosis.88 Although the inflammasome is canonically activated by extracellular PAMP recognition, the inflammasome is also activated via C5a binding intracellularly to the mitochondria, with mitochondrial C5aR2–ROS–NOX2 implicated in activation.89
Inflammasomes play a nuanced role in cancer, and are associated with promoting angiogenesis and immunosuppression in some cancers, such as breast,90 while supporting Th1 and NK cell activity in others, such as CRC.91 In a murine C5aR2 knockdown model, C5aR2 deficiency inhibited NLRP3 activation in vivo, indicating the importance of intracellular complement signalling in inflammasome formation.83
CD46 signalling
4.5
CD46, expressed on all nucleated cells, integrates complosome with T‐cell fate and antitumour response. In its inactivated state, CD46 maintains T‐cell homeostasis by binding to Jagged‐1, shunting Notch signalling and T‐cell differentiation.61 When T cells are activated by the TCR, CD46 is shed, and Notch is induced, triggering Th1 differentiation.61, 92 CD46 also governs resolution of Th1 responses, driving IL‐10 and T‐cell plasticity. When CD46 is activated, IL‐10 is expressed in CD4^+^ cells and Th1 cells, inducing a switch in cytokine production from IFN‐γ to IL‐10.93 Clinically, CD46 is generally overexpressed in cancers, including ovarian, hepatocellular and breast cancers.94 Given its role in IL‐10 induction, dysregulated CD46–IL‐2R/Notch axes may contribute to tumour immune evasion.
COMPLOSOME AND IMMUNE CELL DYNAMICS
5
Regulatory T cells
5.1
Regulatory T cells (Tregs) are thought to be in part regulated by the complosome and complement proteins, and play important roles in maintaining self‐tolerance (Figure 4). However, in cancer, Tregs promote tumorigenesis via the production of suppressive cytokines (TGFβ and IL‐10) and stimulation of angiogenesis via the secretion of VEGF.95 Many tumour tissues are enriched in immunosuppressive Tregs, and having a higher proportion of Tregs is associated with worse overall survival and risk of recurrence in multiple cancers, including CRC, NSCLC and triple‐negative breast cancer (TNBC).96, 97, 98, 99, 100, 101, 102
Overview of immune mechanisms regulated by intracellular complement proteins. In T cells, TAMs and Tregs, complosome signalling drives metabolic reprogramming, inflammation and immune suppression. Neutrophils and MDSCs are activated to support chemotaxis and pro‐tumour signalling, while NK cells’ cytotoxic function is inhibited. Additionally, CAFs are primed by intracellular C3 to secrete factors including CXCL12 and TGFβ, enhancing immune evasion by shunting T‐cell expansion and promoting tumour growth.
Higher numbers of FOXP3^+^ Tregs were observed in C3‐deficient mice, suggesting C3 may contribute to blocking Treg differentiation.103 Elevated Treg populations have also been observed in systemic lupus erythematosus (SLE) patients with lower plasma levels of C3 and C4.104 Interestingly, complement C4 protein has been shown to induce Treg differentiation directly in an in vitro model, where C4 was co‐cultured with T cells and DCs isolated from patients with SLE.105 While C3 and C4 are likely linked to Treg expansion, the mechanisms regulating this association remain largely unknown.
B cells
5.2
B cells can be pro‐ or antitumour depending on the microenvironmental context. High B cell infiltration is often associated with longer survival in breast, ovarian, renal and lung cancers, in part through the formation of tertiary lymphoid structures (TLS) with DCs and T cells that enhance antigen presentation and T‐cell priming.106, 107 TLS are lymphoid aggregates that form de novo in non‐lymphoid tissues in response to chronic inflammation or cancer, and are commonly found within and around cancer tissue.108 Conversely, B regulatory cells (Bregs)—driven by cytokines, such as IL‐21, IL‐6, IL‐33, IL‐35, IL‐1β and IFN‐α, secrete IL‐10 and promote immunosuppression.109, 110, 111
Within this spectrum, the complosome is a core regulator of B‐cell physiology. B cells contain intracellular C3/C3a and can generate C3 autocrinally, which is necessary for B cell self‐renewal and survival; serum‐derived C3 can also be imported and processed via cathepsin L to C3b/C3a, with nuclear entry reported for exogenous C3.9, 10, 112 B cells express C3aR1/C5aR1, and genetic disruption of these receptors abrogates mTOR activation after BCR/CD40 stimulation, leading to premature germinal‐centre collapse and defective maturation—highlighting a requirement for complosome signalling in B‐cell activation.113, 114
Complosome signalling in B cells has also been associated with epigenetic modulation of gene expression. C3a inhibits histone H1 binding to DNA in a dose‐dependent manner.10 In cancer, the knockdown of H1 in breast cancer cells has been associated with altered gene expression, type I IFN production and a hyperproliferative phenotype.115
DCs
5.3
DCs orchestrate antitumour T cell responses through antigen presentation and co‐stimulation, and they express C3aR along with complement regulators CD49, CD55 and CD59.116, 117 Intracellular deficiency of C3 has been associated with impaired DC differentiation; however, the mechanism by which this occurs remains poorly understood.118 DCs have produced and synthesised C3 intracellularly in vivo and in vitro, and C3‐inhibited DCs exhibit reduced ability to stimulate alloreactive T cells, polarise CD4^+^ T cells to a Th2 phenotype and drive Treg expansion.119
Beyond differentiation, the complosome in DCs modulates antigen handling and the tempo of T cell priming. Whereas extracellular opsonisation tags targets via C3b for phagocytosis, DCs themselves display C3 fragments, enabling a form of intracellular opsonisation that shapes antigen processing.120 In apoptotic cell models, C3‐sufficient DCs retained intact cargo (lysosomal debris) longer, indicating that C3 fragments act as chaperones of phagosome maturation. Conversely, C3‐deficient DCs digested cargo rapidly and elicited diminished CD4^+^ proliferation, effectively pacing down the T cell response.121 Unsurprisingly, T cell response was ‘paced’, as DCs deficient in C3 consumed the target antigen faster and elicited diminished CD4^+^ T cell proliferation compared to the C3‐sufficient DCs, which had not digested the apoptotic cell by endpoint and elicited a more robust T cell response.121 These data support a role for intracellular C3 in regulating antigen‐trafficking kinetics and MHC‐II exposure, with potential implications for cancer: reduced DC C3 could truncate antigen presentation and blunt antitumour immunity.
Neutrophils
5.4
As the most abundant innate immune cells in bone marrow and blood, neutrophils have also been associated with promoting cancer progression and poor prognosis when polarised to a tumour‐associated ‘TAN’ state.122, 123 TANs inhibit CD8^+^ T cells and secrete MMP9, VEGF and Arg1.124 Higher neutrophil infiltration in tumour stroma is associated with poorer prognosis in a variety of cancers, including breast, lung and glioma.125, 126 The link between complement and neutrophils was first identified in 1993, when C3 was characterised as a strong chemoattractant for neutrophils in vivo.127 The role of C3 in neutrophil chemotaxis was validated more recently in a murine model of acute pancreatitis, where C3 expression was required for the recruitment of neutrophils to the pancreas.128 As an innate immune cell, neutrophils are known to have potent intracellular stores of C3, and intracellular complement likely mediates neutrophil cytoskeletal maintenance.129 In a study assessing neutrophils from healthy controls and SLE patients, neutrophils from healthy patients exhibited upregulated C3 and were associated with the preservation of cytoskeletal organisation.130 In the context of SLE, cytoskeletal abnormalities impair TCR signalling and immune synapse formation between T cells and APCs, which contribute to disease progression.131 However, in cancer, increased C3 may also promote a pro‐tumour TAN phenotype. In a C3‐knockout murine model of ischaemia‐reperfusion injury, C3 was a requirement for increased neutrophil recruitment to the injury site and neutrophil extracellular trap (NET) formation.132 NETs are composed of condensed nuclear and mitochondrial DNA, proteases and pro‐inflammatory mediators, which may promote EMT and/or activate dormant cancer cells.133
Natural killer cells
5.5
Natural killer (NK) cells limit tumour growth via granzyme/perforin‐mediated cell killing and the production of inflammatory cytokines and chemokines. Like CD8^+^ cells, NK cells can become dysfunctional in the TME as a result of stromal interactions with immunosuppressive cancer‐associated fibroblasts (CAFs), tumour‐associated macrophages (TAMs) and MDSCs, preventing cancer cell killing.134
NK cells highly express C3aR, C5aR, C5aR2, CR3 and CR4; however, it is unknown whether NK cells can secrete or produce C5 and/or C3 intracellularly.135 Interestingly, in a C3‐knockout murine melanoma model, C3 deficiency resulted in increased antitumour immunity of NK cells and reduced tumour growth.136 Depletion of C3 has also been shown to enhance the ability of target cells coated with rituximab to activate NK cells and improve the efficacy of monoclonal antibody therapy in a murine lymphoma model.137 Similarly, C3aR signalling has been shown to inhibit NK infiltration in vivo by triggering the formation of high‐affinity LFA‐1, a critical lymphocyte trafficking integrin, suggesting that C3/C3aR blockade may support anticancer NK responses.138
Macrophages
5.6
Macrophages are early, prevalent TME infiltrates. When polarised to M2‐like (pro‐tumour) TAMs, they secrete immunosuppressive cytokines and ROS, and higher TAM burden predicts poorer outcomes in pancreatic, gastric and breast cancers.139 Higher infiltration of TAMs has been associated with poorer prognosis in numerous cancers, including pancreatic, gastric and breast cancer.140, 141, 142
Intracellular C3 in tumour cells is associated with the promotion of an immunosuppressive macrophage phenotype. Zha et al. demonstrated that intracellular activation of tumour‐derived C3 (resulting in the intracellular generation of C3a and C3b) promoted the generation of TAMs via activation of the C3a–C3aR–PI3K pathway in vivo.143 Importantly, the activation of PI3K has been shown to activate other pro‐tumorigenic pathways in TAMs, such as NF‐κB, which functions to sustain hyperproliferation and immunosuppression.144 This generation of M2‐like macrophages was also associated with the suppression of CD8^+^ T cells, a common interaction between TAMs and T cells. Further, when C3 was deleted in high C3‐expressing tumour‐bearing mice (implanted with 4T1 mammary carcinoma cells), mice were sensitised to anti‐PD1 therapy, and reduced tumour burden was observed.143 The induction of C3 gene transcription can also be activated by the binding of LFA‐1, and LFA‐1 activation may inhibit NK cells and promote M2 macrophages simultaneously.145
Additionally, as previously described in T cells, macrophages and monocytes use autonomous, constitutive and intracellular C3 and C5 activation (engaging with mitochondrial C5aR) to sustain metabolic reprogramming and inflammasome formation.1, 77 As demonstrated by Niyonzima et al., continuously synthesised intracellular C5 shifts ATP production towards ROS generation, aerobic glycolysis and promotes IL‐1β expression in macrophages.77, 146
MDSCs
5.7
MDSCs are another prevalent immune cell population in the TME, implicated in immunosuppression and carcinogenesis. Like Tregs, they restrain CD8^+^ T‐cell expansion via IL‐10, arginase and ROS, and express high levels of C5aR1.49, 147 C5a, generated intracellularly and/or locally within tumours, acts as a potent chemoattractant; once released, it recruits MDSCs to tumour and stromal sites and directly signals via C5aR1 on MDSCs to enhance suppressive function.49, 148, 149 Consistent with local production, C5/C5a is detected within the TME and is secreted by phagocytes.48, 150 In cervical cancer models, C5aR1 sufficiency was required for the intratumoral accumulation of polymorphonuclear MDSCs.48 In colon cancer, anti‐PD‐1/PD‐L1 therapy increased C5a intratumorally, which amplified MDSC inhibitory activity and boosted ROS/NOS/arginase output; C5a/C5aR1 blockade restored antitumour effects of checkpoint therapy.151 Lastly, in hepatocellular carcinoma cells, C3 secretion induced by PIWIL1 gene activation (upregulated in 80.4% of colon cancer cases), activated oncogenic p38 and MAPK signalling in MDSCs.46, 152
Fibroblasts and CAFs
5.8
Cancer‐associated fibroblasts (CAFs) are abundant stromal cells in the TME. Recent work shows that intracellular C3 can be cleaved to generate C3a/C3b, which then signals autocrinally via C3aR1 (endosomal or plasma‐membrane) to drive ‘tissue priming’ and metabolic reprogramming.51, 153, 154 Primed fibroblasts increase pro‐inflammatory IL‐6/IL‐8 and CCL2/CXCL12, switch to aerobic glycolysis, and encode transcriptional/epigenetic memory of past inflammatory events, heightening responsiveness to later stimuli, a phenotype commonly observed in autoimmunity and chronic inflammation, and is increasingly implicated in tumour‐associated stromal activation.155, 156 In synovial models, tissue priming depends on intracellular C3/C3a, drives glycolysis, and activates mTOR and HIF‐1α.154, 157 In the context of cancer, intracellularly generated and secreted C3a can promote cancer invasion and metastasis via the activation of CAFs in murine models.158
Concordant data in gastric cancer models show CAF‐intrinsic C3 associates with NF‐κB upregulation, CD8^+^ dysfunction and therapeutic resistance. Patient samples also revealed an association between increased C3^+^ CAFs in the stroma and stromal infiltration of immunosuppressive M2 macrophages.159 Targeting the NF‐κB/C3 pathway by silencing p65 through siRNA significantly decreased C3 secretion in CAFs; however, the exact intracellular cues that drive initial C3 cleavage are currently unknown.159 Taken together, CAF complosome activation, via intracellular C3 cleavage and autocrine C3aR1/CD46 signalling, drives metabolic rewiring, myeloid recruitment and broad immunosuppression within the TME.51, 154, 156
THERAPEUTIC TARGETING OF THE COMPLOSOME‐IMMUNE AXIS IN CANCER
6
Given its roles in tumorigenesis and immune regulation, the complosome is an attractive therapeutic axis, though no complement‐directed drugs are yet approved in oncology.160, 161 While anticancer complement‐inhibitory drugs are in development, most aim to target extracellular proteins, leaving the intracellular arm relatively unexplored.162
One leading preclinical strategy is C3aR/C5aR blockade combined with traditional immune checkpoint inhibitors. In lung cancer models, C5a–C5aR1 antagonism plus anti‐PD‐1 reduced tumour growth, prolonged survival, decreased MDSCs and restored CD8^+^ effector function.82, 163, 164, 165 In melanoma, co‐delivery of C3aR1/C5aR1 antagonists with anti‐PD‐1 produced synergy beyond PD‐1 alone, replicated across studies.82, 151, 165 It has also been demonstrated that C5aR blockade may reprogramme M2 TAMs and synergise with anti‐PD‐1 therapy.166 In a gastric cancer model, higher intratumoral densities of C5aR1^+^ TAMs, the compartment with the strongest C5aR1 signal, associate with worse overall survival and chemoresistance.167 Selective C5aR1 antagonism reduced tumour proliferation, lowered IL‐6/IL‐10/TGF‐β from TAMs (consistent with M1 repolarisation), and reinvigorated CD8^+^ cytotoxicity in vivo.166
Complement inhibition can also potentiate cytotoxic therapies. Radiotherapy increases intratumoral C3/C3b and C3a/C5a in ovarian tumour models168; In CRC, systemic C5aR1 antagonism improved radiation responses.169 Systemic blockade of C5aR1 has also shown preclinical synergistic effects with chemotherapy. C5aR1 blockade plus paclitaxel enhanced CD8^+^ proliferation/cytotoxicity in an IFN‐γ‐dependent manner in SCC.50 Data from our group link complement to neoadjuvant chemoradiation (nCRT) response across gastrointestinal cancers.170, 171 In oesophageal adenocarcinoma (OAC), it was demonstrated that pretreatment serum levels of complement anaphylatoxins (C3a/C4a) predict poor response to nCRT and adverse outcomes.170 More recently, in rectal cancer, we reported that complement is increased in radioresistant rectal cancer cells and that C3 functionally modulates the radioresponse in vitro, with inhibition of C3 reversing a radioresistant phenotype, concomitant with increased radiation‐induced DNA damage and a shift towards a more radiosensitive cell cycle phenotype.172 Collectively, these findings implicate complement, particularly C3 and its downstream effectors, as a determinant of resistance to cytotoxic therapy and a rational target for radio‐chemo‐immunotherapy combinations.
Therapeutically, translating these preclinical studies into the clinic largely depends on agent selection and receptor selectivity, access to intracellular complement pools and delivery to relevant cell types. Current tools, including C5aR1 antagonists (PMX53, PMX205, avacopan/CCX168) and C3aR1 tool/antagonist compounds (SB290157, context‐dependent pharmacology), primarily engage cell‐surface receptors.165, 173, 174, 175 The dual small‐molecule DF2593A blocks C3aR1/C5aR1 in vitro, and JPE1375 shows activity against C5aR1, including mitochondrial pools in select settings.77, 176 However, selective access to intracellular C3 versus extracellular C3 is limited.1
Despite its therapeutic promise, targeting intracellular complosome signalling presents substantial translational challenges. Key barriers include the difficulty of achieving subcellular delivery to intracellular receptors such as C5aR1,177 tumour microenvironment heterogeneity that may drive variable therapeutic responses, and the risk of unintended immune perturbation due to the pleiotropic roles of complement in host defence.178, 179 Additionally, most available complement inhibitors were developed to neutralise extracellular pathways and may not effectively engage intracellular targets. Emerging strategies to overcome these challenges include tumour microenvironment‐responsive nanocarriers,180 cell‐type‐restricted delivery platforms,181 and biased receptor modulators designed to selectively inhibit pathogenic intracellular signalling while preserving homeostatic immune functions.182 Advances in single‐cell profiling and spatial transcriptomics may further enable rational patient stratification and guide precision targeting of complosome‐driven tumour states.
Beyond therapy, the complosome shows promise as a biomarker. Profiling a patient's intracellular complosome and ‘complotype’ (complement‐gene polymorphisms) may inform prognosis and treatment selection.172, 183 Potential approaches include quantification of intracellular C3 or C5 expression, assessment of C3a/C5a signalling signatures, or integration of complotype variants with transcriptional and metabolic profiling.36, 43 However, standardised detection methods, clinically actionable thresholds, and prospective validation in patient cohorts remain largely unexplored, underscoring the need for translational studies to define the clinical utility of complosome‐based biomarkers.184, 185 While systemic complement and complosome testing are not yet routinely included in oncology patient profiling, given the role of the complosome in cancer and immune regulation, complosome proteins may be important novel biomarkers in prognostic, diagnostic and clinical decision‐making. Prospective evaluation within oncology workflows, especially among cancer types where early diagnosis is challenging, such as OAC, is warranted.
CONCLUSION
7
The complosome represents an emergent axis of tumour‐immune regulation with relevance to cancer progression, treatment resistance and inflammatory comorbidity. In this review, we highlight evidence across preclinical models indicating that intracellular complement shapes T‐cell expansion and differentiation, promotes immunosuppression through MDSC, macrophage and CAF recruitment/polarisation, and reprogrammes metabolism via mTOR activation, glycolytic flux and ROS/NOS. Therapeutic strategies to modulate complosome C3 activity, particularly through C3aR and C5aR antagonism, hold promise in reducing immunosuppression and pro‐tumour metabolic reprogramming in immune cells. Further research is required to unravel the exact mechanisms underlying intracellular C3 cleavage and complosome activation in tumour cells and immune cells. Future research priorities include (i) achieving compartmental specificity (selective access to intracellular vs. extracellular complement); (ii) building pharmacodynamic endpoints (myeloid trafficking; T‐cell metabolism/exhaustion) into study and trial design; and (iii) standardising complement/complosome biomarker panels for routine oncology workflows.
AUTHOR CONTRIBUTIONS
A.B., J.L. and N.L.L. conceptualised the review. A.B. wrote the original draft manuscript and prepared the figures. All authors read and edited the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflicts of interest.
ETHICS STATEMENT
The authors have nothing to report.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1West EE , Kemper C . Complosome—the intracellular complement system. Nat Rev Nephrol. 2023;19(7):426‐439. doi:10.1038/s 41581‐023‐00704‐1 37055581 10.1038/s 41581-023-00704-1PMC 10100629 · doi ↗ · pubmed ↗
- 2Manderson AP , Pickering MC , Botto M , Walport MJ , Parish CR . Continual low‐level activation of the classical complement pathway. J Exp Med. 2001;194(6):747‐756. doi:10.1084/jem.194.6.747 11560991 10.1084/jem.194.6.747PMC 2195964 · doi ↗ · pubmed ↗
- 3Lubbers R , van Essen MF , van Kooten C , Trouw LA . Production of complement components by cells of the immune system. Clin Exp Immunol. 2017;188(2):183‐194. doi:10.1111/cei.12952 28249350 10.1111/cei.12952 PMC 5383442 · doi ↗ · pubmed ↗
- 4Khan MA , Assiri AM , Broering DC . Complement and macrophage crosstalk during the process of angiogenesis in tumor progression. J Biomed Sci. 2015;22(1):58. doi:10.1186/s 12929‐015‐0151‐1 26198107 10.1186/s 12929-015-0151-1PMC 4511526 · doi ↗ · pubmed ↗
- 5Liszewski MK , Kolev M , Le Friec G , et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143‐1157. doi:10.1016/j.immuni.2013.10.018 24315997 10.1016/j.immuni.2013.10.018PMC 3865363 · doi ↗ · pubmed ↗
- 6Chen S , Zhang J , Chen J , et al. Compromised C 3b‐VSIG 4 axis between decidual NK cells and macrophages contributes to recurrent spontaneous abortion. J Transl Med. 2024;22(1):1017. doi:10.1186/s 12967‐024‐05829‐w 39529122 10.1186/s 12967-024-05829-w PMC 11556194 · doi ↗ · pubmed ↗
- 7Hess C , Kemper C . Complement‐mediated regulation of metabolism and basic cellular processes. Immunity. 2016;45(2):240‐254. doi:10.1016/j.immuni.2016.08.003 27533012 10.1016/j.immuni.2016.08.003PMC 5019180 · doi ↗ · pubmed ↗
- 8Reichhardt MP , Meri S . Intracellular complement activation—an alarm raising mechanism? Semin Immunol. 2018;38:54‐62. doi:10.1016/j.smim.2018.03.003 29631809 10.1016/j.smim.2018.03.003 · doi ↗ · pubmed ↗