Increased prevalence of coronary heart disease among current smokers carrying APOL1 risk variants within the African American population
Jelena Mustra Rakic, Clive R. Pullinger, Erin L. Van Blarigan, Irina Movsesyan, Eveline Oestreicher Stock, Mary J. Malloy, John P. Kane

TL;DR
African American smokers with certain APOL1 gene variants may have a higher risk of heart disease, especially if they carry two copies of the variant.
Contribution
This study identifies an interaction between APOL1 risk variants and smoking in increasing coronary heart disease risk in African Americans.
Findings
Current smokers with APOL1 risk variants had 3.3 times higher odds of CHD compared to nonsmokers.
Those with two APOL1 risk alleles had 7.3 times higher odds of CHD compared to nonsmokers.
Former smoking was associated with CHD regardless of APOL1 genotype.
Abstract
The apolipoprotein L1 (APOL1) G1 and G2 gene variants, highly prevalent among the African American population (rare in other racial groups), are linked to increased risk of kidney disease, sepsis, and potentially coronary heart disease (CHD). Their role in tobacco-related CHD remains unclear. To investigate the effect of APOL1 risk variants on the association between tobacco smoking and prevalent CHD in African American adults. We conducted a cross-sectional study involving 519 African American adults recruited through the University of California San Francisco Lipid Clinic. Using multivariable logistic regression, we assessed the association between tobacco smoking and CHD, overall and with its most severe subtype, myocardial infarction (MI), among all participants and APOL1 genotype subgroups. Among participants, 41% were current (14%) or former (27%) smokers, 54% carried APOL1…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
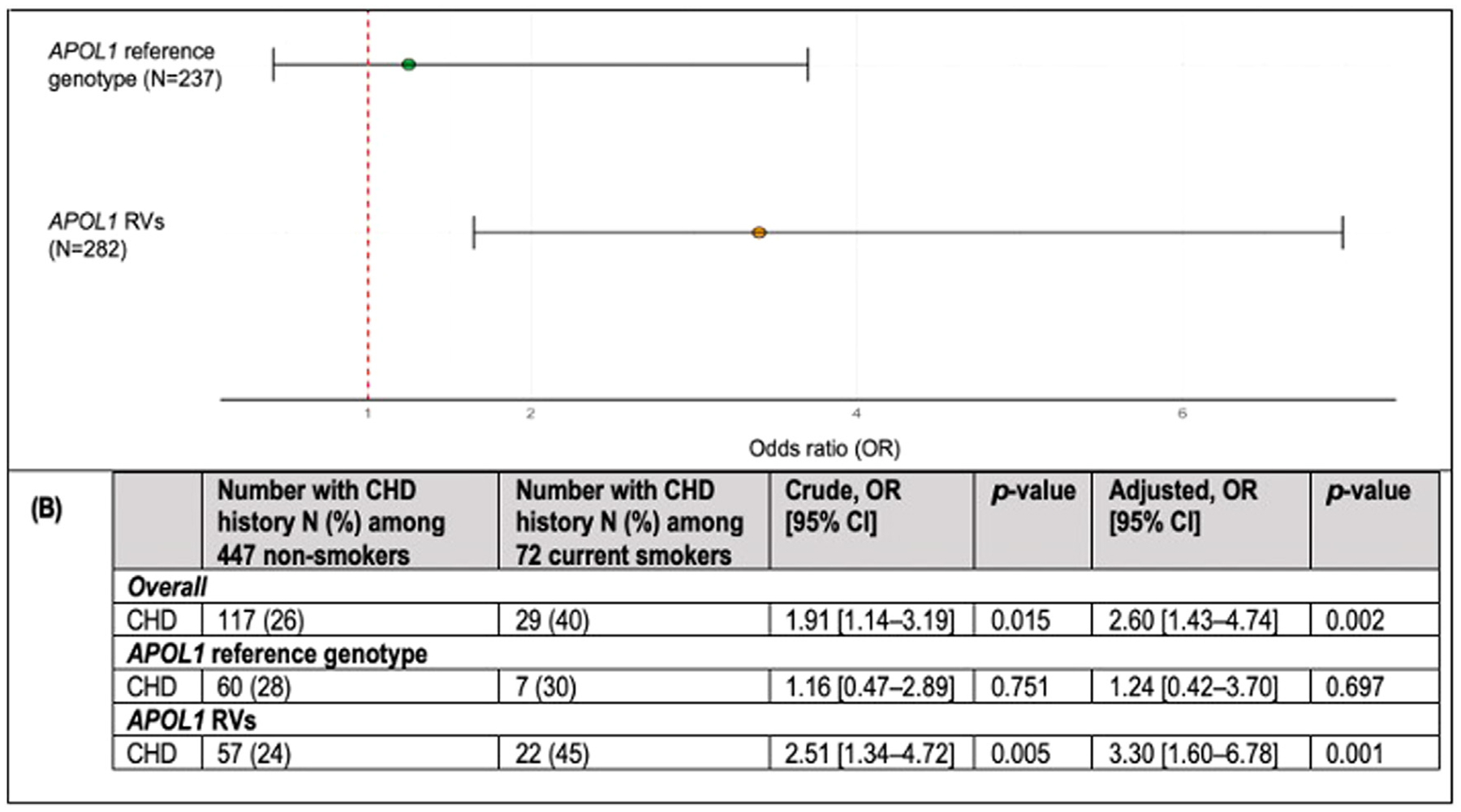 Figure 1
Figure 1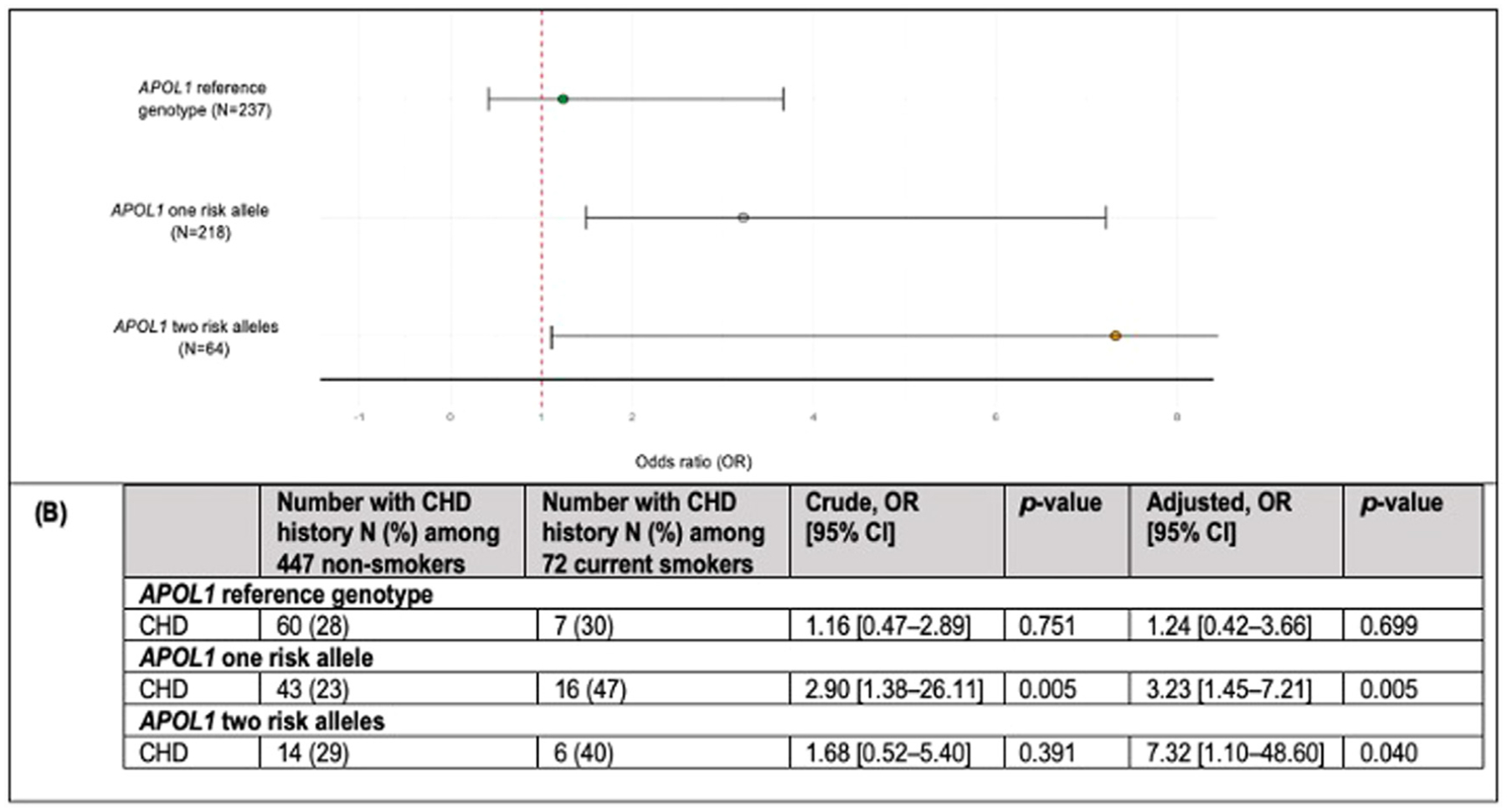 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Paraoxonase enzyme and polymorphisms · Diabetes Management and Research
Introduction
African American individuals bear a high burden of coronary heart disease (CHD), with age-adjusted mortality rates exceeding those of other racial and ethnic groups in the United States.^1–3^ Despite ongoing efforts to reduce cigarette smoking, a well-established risk factor for CHD, African Americans continue to have the lowest quit rates compared to Hispanics/Latinos and non-Hispanic Whites.^4^ Recent findings indicate that smoking disproportionately increases CHD risk in African Americans.^5^ Even after accounting for traditional risk factors like obesity, diabetes, hypertension, and socioeconomic status, African American smokers still face a higher risk of developing tobacco-related CHD compared to White smokers.^5^
The apolipoprotein L1 (APOL1), found in high-density lipoprotein (HDL) and highly expressed in the vasculature, protects against Trypanosoma brucei, the parasite responsible for African sleeping sickness.^6–9^ Two rare variants (RVs) exist in the C-terminus: G1 and G2.^6^ G1 consists of 2 mutations (S342G and I384M) in linkage disequilibrium. G2 involves a 2-amino acid deletion (del388N389Y).^6,7,10^ Both provide extended protection against African Trypanosoma brucei.^11,12^ Homozygosity or compound heterozygosity is strongly associated with increased risk of kidney disease, sepsis, and potentially cardiovascular disease in the African American population.^6,10,13–17^
Findings on the link between these RVs and CHD are inconclusive.^17,18^ Evidence indicates a strong need to understand the role of other risk factors surrounding APOL1 RV-associated diseases. The second hit hypothesis suggests that the presence of APOL1 RVs alone, particularly a single risk allele, may be insufficient to trigger disease. Additional factors, such as inflammatory insults, may be required to increase APOL1 expression to a critical threshold that initiates disease onset.^13,18–20^ APOL1 RV carriers exhibit endothelial cell dysfunction^13^ and increased risk of thrombotic coronary death, the latter linked to APOL1 protein accumulation in the necrotic core and increased plaque instability.^21^ Smoking elevates systemic inflammation,^22,23,24^ which can increase APOL1 gene expression.^25^ Tobacco-related inflammation may act as a second hit by upregulating APOL1 gene expression, potentially exacerbating endothelial dysfunction and necrotic core enlargement, ultimately increasing CHD risk in smokers with APOL1 RV.
Approximately 50% of African American people carry at least 1 APOL1 risk allele.^6,16,17,19^ These RVs are exceedingly rare in other racial groups. Given this high frequency of RVs and the proinflammatory effects of smoking, we aimed to determine if the presence of these RVs modifies the relationship between tobacco smoking and CHD prevalence in African American adults.
Materials and methods
Study design and participants
We conducted a retrospective cross-sectional study using samples and data from participants in the University of California, San Francisco (UCSF) Genomic Resource in Arteriosclerosis (GRA) study, which adhered to the World Medical Association Declaration of Helsinki and was approved by the UCSF Institutional Review Board as part of the UCSF Human Research Protection Program. The GRA includes a large repository of DNA and plasma samples with clinical data, collected over >20 years. Participants were recruited through the UCSF Lipid Clinic, where individuals with dyslipidemia, CHD, diabetes, obesity, other metabolic concerns, or a family history of these conditions were referred. All participants provided written informed consent prior to enrollment. The proportion of African Americans recruited is consistent with their representation in the Bay Area population (approximately 7%^26^). For our study, we included only those who self-identified as African American, were 18 years or older, and had available clinical data and DNA samples. Of 527 identified African American adults, 519 were eligible for analysis (8 were excluded due to missing CHD history). Data are available upon reasonable request.
Ascertainment of CHD, APOL1 genotype status, and smoking status
Histories of CHD were obtained as before.^27^ Briefly, patients were considered to have CHD if they met any of these criteria: history of myocardial infarction (MI), evidence of coronary disease on angiogram, previous stent placement, angioplasty, or coronary revascularization. Angiographic disease was graded by a “sum score” (sum of percent luminal intrusion of all lesions). A sum score of 60 was the threshold discriminant for diagnosis of CHD.^27^ MI was determined based on specific criteria: elevated troponin levels, electrocardiographic evidence, and/or echocardiographic studies.
Blood was collected after overnight fasting in tubes containing 0.1% ethylenediaminetetraacetic acid and was centrifuged at 1000 g for 15 minutes at 4 °C to separate plasma. Plasma was stored at −80 °C. Genomic DNA was extracted using the Wizard purification kit (Qiagen) and APOL1 genotypes determined by Sanger sequencing as before.^19^ Participants were grouped into APOL1 RV group (1 or 2 risk alleles: G1/G0, G2/G0, G1/G1, G2/G2, G1/G2) or a reference group (no risk alleles: G0/G0).
Smoking status was obtained from the questionnaire and validated using plasma nicotine and cotinine levels measured at the UCSF Tobacco Biomarkers Core Facility as previously.^19^ This identifies active smoking but lacks sensitivity to detect secondhand smoke exposure (eg, plasma cotinine level greater than 5.92 ng/mL classifies current smokers among African American individuals).^28^ We compared self-reported smoking status with nicotine and cotinine levels (quantitation limits: 1 ng/mL for nicotine, 10 ng/mL for cotinine). Participants with detectable cotinine and/or nicotine were reclassified, where appropriate, as current smokers. Smoking history was assessed by classifying those not identified as current smokers as nonsmokers. Nonsmokers were further categorized as former smokers if they had a history of smoking, or as never smokers if they did not.
Clinical and lifestyle covariates
Plasma levels of total cholesterol (TC), HDL cholesterol (HDL-C), and triglyceride (TG) were measured as previously.^29^ Hypertension, type 2 diabetes mellitus, and dyslipidemia were defined as before.^19^ Being physically active was defined if participants reported exercising ≥30 minutes (including walking) more than twice per week, and alcohol intake was defined as consuming >2 alcoholic drinks/week. To assess kidney function, we measured plasma creatinine (mg/dL) (QuantiChrom Kit, BioAssay Systems, Hayward, CA). Using creatinine levels, age, and sex (defined as sex assigned at birth), estimated glomerular filtration rate (eGFR) was calculated; eGFR<60 mL/min/1.73 m^2^ indicated mild to moderate kidney function loss.
Statistical analysis
Power calculation was based on data from a previous similar study.^5^ The odds ratio (OR) among carriers was expected to exceed that for the overall African American population. With an alpha of 0.05 and 80% power, 255 participants with APOL1 RVs were sufficient to detect an OR>2.8 for the association between smoking and CHD among carriers. Statistical analyses were conducted using R (version 4.4.1) software. Statistical significance was determined at α < 0.05 (2-sided). Variables were checked for missing values and skewness. Participants’ characteristics were summarized by smoking status. P-values and descriptive statistics are presented as mean ± SD or median (range) for continuous variables and as frequencies (%) for binary and categorical variables. Comparisons of distributions were performed by unpaired t-test for continuous variables or Pearson χ^2^ test for categorical variables.
To examine the relationship between smoking status and CHD, we used Firth’s logistic regression (‘logistf’ R package), categorizing smoking status as current vs nonsmokers (former and never smokers). Adjustments included age, sex, dyslipidemia, hypertension, diabetes, body mass index (BMI), and eGFR. Missing data were addressed using the “MICE” package, employing predictive mean matching (pmm) assuming data were missing at random. Due to significant missing data for alcohol and exercise (>30% of participants had missing values) these variables were omitted from the main model. Sensitivity analyses, including these variables among participants with complete data, showed no substantial impact on the magnitude of the association between smoking and CHD prevalence. To test for effect modification of the association between smoking and CHD history by APOL1 genotype, an interaction term between the dichotomous smoking variable and APOL1 genotype (yes/no) was included in multivariate models. In our secondary analysis, which was exploratory in nature, we examined the relationship between smoking and CHD separately among APOL1 reference genotype subgroup, carriers of 1 risk allele, and carriers of 2 risk alleles (additive model). We also analyzed the association between current vs nonsmokers and prevalent MI stratified by APOL1 genotype (dominant model).
In a sensitivity analysis, we assessed the association between smoking status (current, former, never) and CHD prevalence or MI, stratified by APOL1 genotype, using a dominant model. In an additional exploratory analysis we performed a t-test to compare the average age of nonsmokers and current smokers with a history of CHD, overall and stratified by APOL1 genotype, to assess potential differences in age of CHD onset.
Results
Participants’ characteristics
Participants’ characteristics are shown in Table 1; 72 (14%) were classified as current smokers, and 447 (86%) current nonsmokers, including 142 (27%) former, and 305 (59%) never smokers. The average age was 57.3 ± 14.0 years (18–88 years). Nonsmokers were older than current smokers (58.2 ± 14.2 years vs 52.9 ± 11.3 years), with slightly more women in the nonsmoker group (53%) and men in the current smoker group (55%). Median BMI was 27 kg/m^2^ (24–32 kg/m^2^), with no marked difference between nonsmokers and current smokers. There were also no significant differences between nonsmokers and current smokers concerning prevalence of diabetes, hypertension, or dyslipidemia. Nonsmokers included a higher proportion of physically active individuals (56% vs 13%) and fewer reported drinking >2 alcoholic drinks/week (42% vs 27%) when compared to current smokers. There were 146 participants with a history of prevalent CHD, of whom 29 (40%) were current smokers and 117 (26%) were nonsmokers. Of the 146 patients with CHD, 8 had insufficient information to determine MI status. Among the 138 CHD patients with available MI data, 82 (59%) had a history of MI. Additionally, 282 (54%) participants carried APOL1 RVs (1 or 2 risk alleles), with 64 (12%) having 2 risk alleles and 218 (41%) having 1 risk allele. The APOL1 G1 (rs73885319, p = .3126; rs60910145, p = .353) and the G2 variant (rs71785313, p = .3832) were consistent with Hardy-Weinberg equilibrium.
Prevalent CHD and MI in current and nonsmokers differed by APOL1 genotypes
Carriers of the APOL1 reference genotype showed a similar prevalence of CHD among current smokers and nonsmokers, 60 (28%) and 7 (30%), respectively, with a similar trend for MI. Among carriers of RVs, current smokers had a significantly higher prevalence of CHD compared to nonsmokers, (22 [45%] vs 57 [24%]) (Fig 1B) and had a significantly higher prevalence of MI compared to nonsmokers (14 [29%] vs 32 [14%]) (Table 2).
APOL1 RVs associated with higher prevalence of CHD and MI among current smokers
The p-interaction suggested that the relationship between smoking and prevalent CHD may vary by APOL1 genotype (p = .074), so we stratified the data by genotypes (Fig 1A,B).
Among 282 APOL1 RV carriers, current smokers had 3.30 times the odds of having a history of CHD compared to nonsmokers (95% CI: 1.6, 6.8, p = .001). In contrast, current smoking was not significantly associated with a history of CHD among 237 individuals with the APOL1 reference genotype (OR = 1.24; 95% CI: 0.4, 3.7, p = .697). A secondary analysis investigating the effect of genotype on the relationship between smoking status (current vs nonsmoker) and prevalent MI, was consistent with that for CHD (Table 2). The association between smoking and MI differed by genotype, with a significant association observed only among APOL1 RV carriers (OR = 3.29; 95% CI: 1.5, 7.4).
Additive effect of APOL1 risk alleles on CHD prevalence among current smokers
To assess whether the presence of 2 APOL1 risk alleles had a stronger effect on the association between smoking and CHD than a single risk allele, we conducted secondary analyses in 3 subgroups (APOL1 reference, 1 risk allele, and 2 risk alleles). The OR for CHD among current smokers compared to nonsmokers increased with the number of risk alleles (0, 1, or 2), with ORs (95% CI) of 1.24 (.4, 3.7), 3.23 (1.5, 7.2), and 7.32 (1.1, 48.6), respectively (Fig 2A,B).
Smoking’s association with CHD and MI is independent of APOL1 genotype in former smokers
In the sensitivity analysis, current smokers among APOL1 RV carriers had 3.63 times the odds of CHD compared to never smokers (95% CI: 1.7, 7.8, p = .001). The association between smoking and CHD was not significant for non-carriers (Table 3). Former smokers with RVs had 1.80 times the odds of having prevalent CHD compared to never smokers; this association did not reach statistical significance (95% CI: 0.9, 3.5, p = .090). A similar relationship was observed among non-carriers (OR = 2.01, 95% CI: 0.9, 4.3, p = .080). Among RV carriers, current smokers had 4.89 times the odds of MI history compared to never smokers (95% CI: 2.0, 12.0, p = .001, Table 4). The association was significant for former vs never smokers (OR = 2.66, 95% CI: 1.2, 5.7, p = .020). Among non-carriers, the relationship between past smoking and MI was significant (OR = 2.40, 95% CI: 1.0, 5.5, p = .043), whereas this was not observed for current smoking and MI (OR = 1.91, 95% CI: 0.6, 6.6, p = .331).
Smokers with APOL1 risk variants may have earlier onset of CHD
Among participants with CHD history, current smokers were younger than nonsmokers; average age 57 years compared to 63 years (p = .018). Within the APOL1 reference group, among those with prevalent CHD, the average age was similar between current smokers and nonsmokers (63 vs 63 years). For carriers of RVs, current smokers with prevalent CHD were significantly younger than nonsmokers; average age 55.5 years compared to 62 years (p = .017) (Table S1).
Discussion
In this well-characterized cohort of African American adults, APOL1 genotype status modified the association between current cigarette smoking and the odds of CHD, including MI. Among individuals with APOL1 RVs, current smokers had 3.3 times the odds of CHD compared to nonsmokers, after adjusting for traditional risk factors. The effect was strongest in carriers of 2 risk alleles (OR = 7.3) and substantial for those with 1 risk allele (OR = 3.2), implying additive effect of these alleles. Among non-carriers, smokers had 1.2 times the odds of CHD, but this was not statistically significant. Given that approximately half of African Americans carry an APOL1 risk allele, these findings highlight a potential interplay between genetic susceptibility and smoking that could contribute to the elevated rates of tobacco-related CHD in this population.
The concept of a second hit in the development of APOL1 RV-associated diseases has been suggested in many studies.^18–20,30,31^ APOL1 RVs are strongly linked to the excess burden of kidney disease among African Americans.^10,13–15^ In a mouse study, kidney disease development in carriers of APOL1 RVs depended not only on the presence of G1 and G2 variants but also on their expression levels.^18^ In animal models and cell cultures, APOL1-induced toxicity was influenced by both the presence of RVs and expression levels, with higher APOL1 mRNA or protein expression associated with increased toxicity.^18,32^ Moreover, human kidney glomerular samples show that elevated APOL1 transcript levels are linked to kidney disease and correlate with kidney function.^18,32^ Inflammatory cytokines, such as IFN-γ and TNF, enhance APOL1 gene expression in human umbilical vein endothelial cells (HUVECs),^11,33^ and even more so in human coronary artery endothelial cells (HCAECs), compared to podocytes.^9^ Studies investigating the relationship between APOL1 RVs and CHD have yielded mixed results.^17,21,34–37^ Variations in cohort characteristics and the presence of additional risk factors, such as proinflammatory conditions, may alter the influence of APOL1 RVs on CHD outcomes. As reported by Gutierrez et al.,^16^ hazard ratios (HRs) for the association between APOL1 RVs and CHD under recessive and dominant models in nondiabetics indicate that a single risk allele does not increase CHD risk. Among diabetics, HR increased in the dominant model.^16^ Although these results were not statistically significant, the trend suggests that even 1 APOL1 risk allele may elevate CHD risk, potentially due to diabetes-related inflammation. This implies that an additional insult is particularly relevant for disease development in carriers of 1 risk allele; carrying 2 may be sufficient to cause disease and the presence of additional insults may further exacerbate the condition. In our study, we observed a significant association between smoking and prevalent CHD among carriers of APOL1 RVs, while the association did not reach significance among non-carriers. The effect of smoking was strongest in carriers of 2 risk alleles (OR = 7.3), followed by carriers of 1 (OR = 3.2), and less pronounced in non-carriers (OR = 1.2). It is important to note that the subgroup of smokers with 2 APOL1 risk alleles was small, resulting in a wide 95% CI and limiting the precision of the effect size estimate. Thus, these findings should be interpreted with caution. Still, the results suggest that the effect is greater in individuals with 2 risk alleles compared to those with only 1, providing valuable insights that warrant further investigation in larger, more statistically powered studies. Overall, these results support the hypothesis that a second hit is important in APOL1 RV-associated CHD development, with smoking, characterized by systemic inflammation, potentially acting as this second hit in RV carriers, leading to increased vulnerability to CHD possibly consistent with an additive inheritance model.
Pathologies associated with APOL1 RVs are linked to endothelial cell dysfunction, with several studies highlighting the critical role of these cells in the disease development.^11,13–15^ Endothelial cells in vessels form the inner lining of the circulatory system with their dysfunction often preceding the onset of cardiovascular disease.^38^ Recently, APOL1 RV carriers were found to have increased levels of adhesion molecules (vascular cell adhesion molecule-1 [VCAM-1] and intercellular adhesion molecule-1 [ICAM-1]) and a proinflammatory phenotype in kidney endothelial cells leading to their dysfunction.^13^ Endothelial cell dysfunction could lead to atherosclerosis and coronary plaque enlargement by increasing monocyte recruitment into vessel walls, their differentiation into macrophages, and greater low-density lipoprotein uptake. In transgenic mice, APOL1 RVs were found to promote cholesterol accumulation in macrophages by downregulating key transporters involved in reverse cholesterol transport, leading to foam cell formation and growth of the necrotic core and plaque.^39^ Cornelissen et al.^21^ found APOL1 RVs increased the risk of thrombotic coronary death due to plaque rupture.^21^ Specifically, individuals carrying risk alleles showed higher APOL1 protein accumulation within coronary plaques and larger necrotic core areas compared to non-carriers. In our study, the association between smoking and increased odds of prevalent CHD, including its most severe subtype, MI, was found to be strong and significant among carriers of APOL1 RVs, but nonsignificant among non-carriers. APOL1 RVs may contribute to the development of tobacco-related CHD by exacerbating the atherosclerotic processes in carriers. Indeed, tobacco smoking is known to elevate inflammatory markers in the circulation,^22,23^ and because endothelial cells are directly exposed to blood, RV carriers may experience heightened endothelial dysfunction and accelerated atherosclerosis, leading to increased tobacco-related CHD risk. Further experimental studies are required to confirm this hypothesis.
To isolate the effect of current smoking on CHD in the context of APOL1 RVs, we classified current smokers using plasma biomarker data, allowing for a detailed assessment of current smoking status. However, the inclusion of former smokers in the nonsmoker group may have attenuated the observed association, particularly if residual risk persisted among recent quitters. This challenge arises from the self-reported nature of former smoking status, which lacked information on cessation timing. The prospective Jackson Heart Study, followed a large cohort of African Americans. CHD risk was significantly higher in current smokers (HR = 2.1) compared to never smokers and elevated for former smokers (HR = 1.4). In a sensitivity analysis we compared former and current smokers. The ORs for CHD were 2.0 for former and 3.3 for current smokers, highlighting the substantial adverse impact of continued smoking and the lower but still notable risk among former smokers. While the association between current smoking and CHD differed by APOL1 genotype, with a strong positive association observed only in carriers, the association between former smoking and CHD did not vary by the genotype. This suggests that, though smoking has lasting effects on cardiovascular health, smoking cessation remains critically important, particularly for carriers of APOL1 RV. As the duration of cessation significantly influences CHD risk—with longer abstinence consistently associated with greater risk reduction—future prospective studies with detailed data on historical smoking exposure, including the timing and duration of cessation, are warranted to understand better the potential interaction between past smoking behavior, APOL1 RVs, and CHD risk.
Among smokers, most CHD cases across all age groups, regardless of sex, can be attributed to smoking.^24,40^ However, compared to never smokers, the relative risk of CHD among current smokers decreases with age.^40^ In a pooled data set of 192,067 women, the HR for CHD among current smokers aged 40 to 49 was 8.5 (95% CI: 5.0, 14), while for those over 70, it was 3.1 (95% CI: 2.0, 4.9).^40^ This suggests that either people susceptible to the adverse impact of smoking develop CHD earlier and/or die, or that the presence of other factors contribute more significantly to CHD risk as people age, reducing the smoking’s relative impact over time. Nevertheless, this implies a younger average age of CHD onset among smokers. Our findings align with this pattern, as smokers with prevalent CHD were, on average, younger than nonsmokers, particularly within the APOL1 RV group highlighting smoking’s pronounced impact in RV carriers. In contrast, no significant age difference was observed between smokers and nonsmokers among non-carriers, possibly reflecting a smaller effect size or limited sample size. Our study design makes it difficult to determine whether the earlier CHD onset in RV carriers who smoke is a true phenomenon or influenced by survival bias. Future studies should use longitudinal data to accurately capture CHD onset in smokers with APOL1 RV.
Our study has several limitations. First, the population was limited to participants who self-identified as African Americans, which restricts the generalizability of our findings to other racial or ethnic groups. However, this focus is justified, as APOL1 RVs are almost exclusively found only in individuals of African descent. Second, due to the cross-sectional observational nature of our study, we are unable to establish causality between the variables examined. Further, it is possible that the associations we observed were in part due to associations of the exposures on the duration of CHD, since we were limited to prevalent CHD events. Additionally, we cannot entirely rule out residual confounding, such as socioeconomic status. Recognizing socioeconomic differences between racial groups^41^ and aiming to minimize the potential confounding effect of socioeconomic status—given that socioeconomic status may contribute to increased systemic inflammation^42^—we included only individuals who self-identified as African American in the study. Third, the lack of a significant association between smoking and CHD for non-carriers may reflect insufficient study power to detect a smaller effect size, and larger studies may be needed. Fourth, in the primary analysis, we applied a dominant model. While this approach does not account for a potential additive effect, emerging evidence suggests that even a single risk allele may increase susceptibility to CHD in smokers. A secondary analysis examined the association between smoking and CHD separately for non-carriers, carriers of 1 risk allele, and carriers of 2 risk alleles. This provided a more nuanced understanding of how the number of risk alleles influences CHD risk in the context of smoking.
In conclusion, this study is the first to examine the role of APOL1 RVs in CHD among African American smokers, utilizing plasma smoking biomarkers to validate self-reported smoking status. With over 500 African American participants, our findings highlight the potential role of genetic susceptibility, specifically the presence of APOL1 RVs, in modulating the impact of tobacco smoking on the prevalence of CHD, including MI. We observed a strong association between smoking and a history of CHD among carriers of RVs, with a dose-dependent increase in the odds of CHD corresponding to the number of risk alleles. While highlighting the overall harm of tobacco smoking, this study underscores its particularly severe adverse impact on individuals with APOL1 RVs, which are highly prevalent among African Americans. These findings have significant public health implications, emphasizing the need for increased awareness and tailored smoking cessation programs within this historically marginalized community. They underscore the importance of personalized healthcare strategies that integrate genetic factors into risk assessments and offer insights into molecular targets for developing targeted interventions.
Supplementary Material
Supplementary Material
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.jacl.2025.04.189.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kyalwazi AN, Loccoh EC, Brewer LC, Disparities in cardiovascular mortality between Black and White adults in the United States, 1999 to 2019. Circulation. 2022;146(3):211–228. doi:10.1161/CIRCULATIONAHA.122.060199.35861764 PMC 9310198 · doi ↗ · pubmed ↗
- 2U.S. Department of Health and Human Services Office of Minority Health. (https://minorityhealth.hhs.gov/omh/browse.aspx?lvl=4&lvlid=19). Accessed September 13, 2024.
- 3Mensah GA. Cardiovascular diseases in African Americans: fostering community partnerships to stem the tide. Am J Kidney Dis. 2018;72(5 Suppl 1):S 37–S 42. doi:10.1053/j.ajkd.2018.06.026.30343722 PMC 6200348 · doi ↗ · pubmed ↗
- 4Nguyen-Grozavu FT, Pierce JP, Sakuma KK, Widening disparities in cigarette smoking by race/ethnicity across education level in the United States. Prev Med. 2020;139:106220. doi:10.1016/j.ypmed.2020.106220.32693179 PMC 7494609 · doi ↗ · pubmed ↗
- 5Oshunbade AA, Kassahun-Yimer W, Valle KA, Cigarette smoking, incident coronary heart disease, and coronary artery calcification in Black adults: the Jackson Heart Study. J Am Heart Assoc. 2021;10(7):e 017320. doi:10.1161/JAHA.120.017320.33754833 PMC 8174312 · doi ↗ · pubmed ↗
- 6Limou S, Nelson GW, Kopp JB, Winkler CA. APOL 1 kidney risk alleles: population genetics and disease associations. Adv Chronic Kidney Dis. 2014;21(5):426–433. doi:10.1053/j.ackd.2014.06.005.25168832 PMC 4157456 · doi ↗ · pubmed ↗
- 7Friedman DJ, Pollak MR. Apolipoprotein L 1 and kidney disease in African Americans. Trends Endocrinol Metab. 2016;27(4):204–215. doi:10.1016/j.tem.2016.02.002.26947522 PMC 4811340 · doi ↗ · pubmed ↗
- 8Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009;19(5):850–858. doi:10.1101/gr.085647.108.19299565 PMC 2675973 · doi ↗ · pubmed ↗