Genome-wide identification and expression profiling of the NHX gene family in oat (Avena sativa L.)
Yuqi Li, Meifeng Liu, Wenjie Zhao, Feng Yuan, Zhenyu Jia, Yi Wang, Wendong Zheng, Chengchen Pan, Chunxiang Fu

TL;DR
This study identifies and analyzes the NHX gene family in oat, revealing their roles in salt stress tolerance and providing insights for improving crop resilience.
Contribution
First comprehensive genome-wide characterization of the NHX gene family in hexaploid oat cultivars.
Findings
126 NHX genes were identified and grouped into 33 orthologous gene groups in four oat cultivars.
Transcriptome data confirmed several AsNHX genes are up-regulated under salt stress, indicating their role in salinity tolerance.
Phylogenetic analysis classified AsNHX genes into three classes: vacuolar, endosomal, and plasma membrane.
Abstract
Salt stress imposes osmotic imbalance and ion toxicity, severely impairing crop growth and development. Na⁺/H⁺ antiporters (NHXs) play a central role in salt tolerance by mediating sodium transport across cellular membranes. We conducted a genome-wide characterization of the NHX gene family in four hexaploid oat cultivars, identifying 126 NHX genes. These were grouped into 33 orthologous gene groups (OGGs), comprising 10 core and 23 dispensable OGGs. The naked oat cultivar ‘Sanfensan (SFS)’ contained the largest number of both total and core NHX genes. Phylogenetic analysis classified AsNHX genes into three classes: vacuolar (Vac-class), endosomal (Endo-class), and plasma membrane (PM-class). Ka/Ks analysis indicated strong purifying selection across most members. Cis-regulatory element analysis revealed abundant stress-related motifs, including abscisic acid (ABA) and methyl jasmonate…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
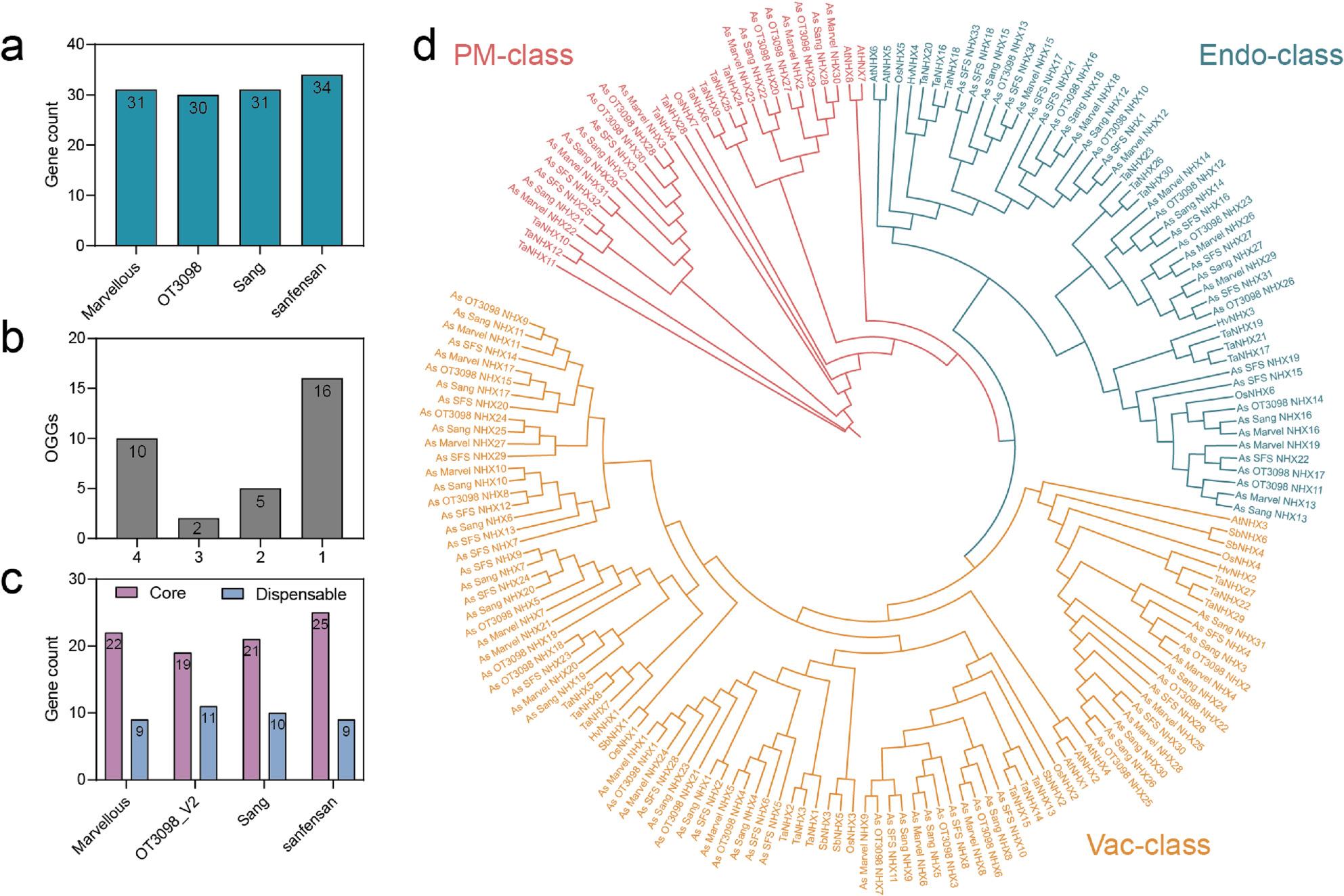 Figure 1
Figure 1 Figure 2
Figure 2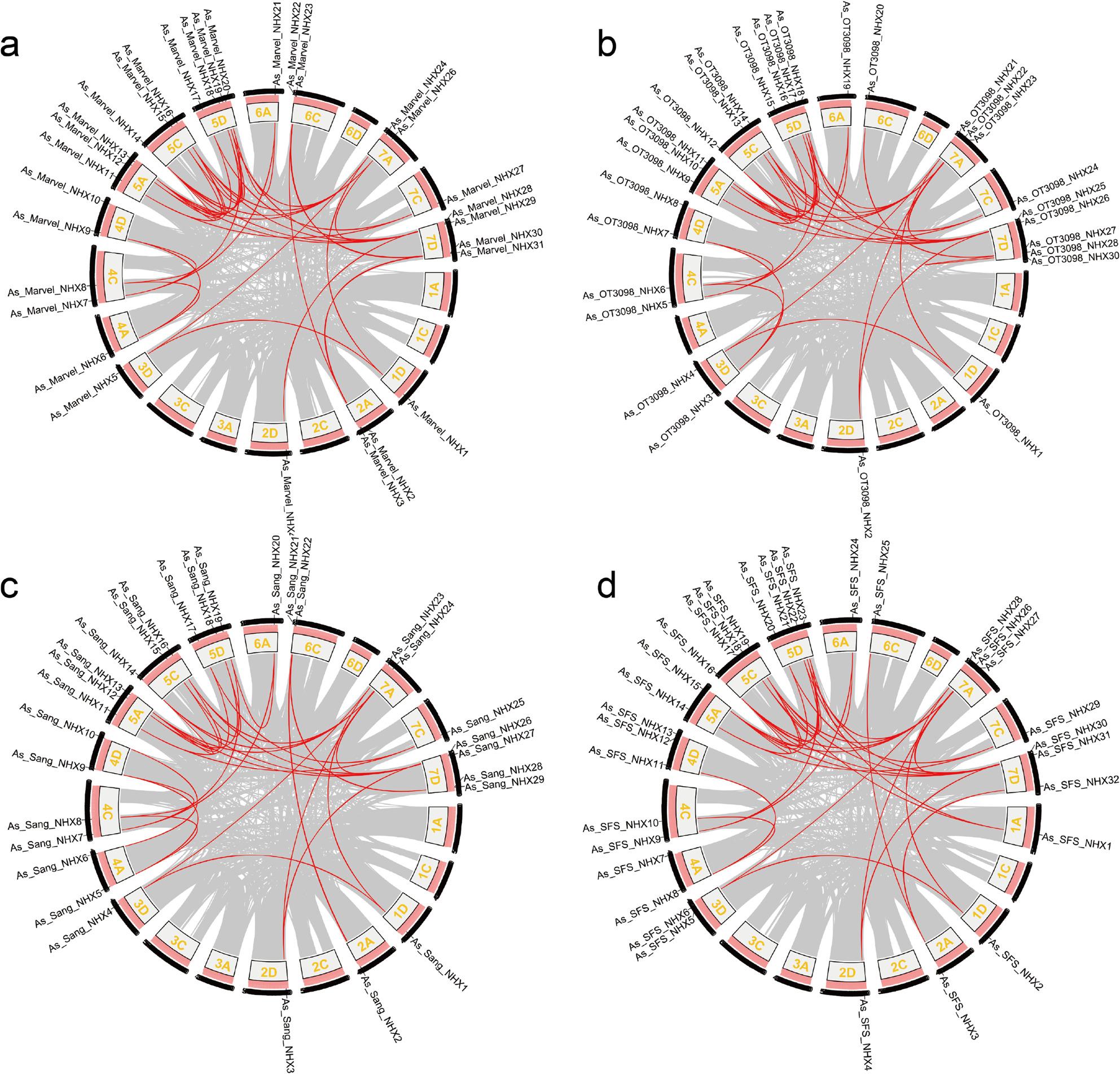 Figure 3
Figure 3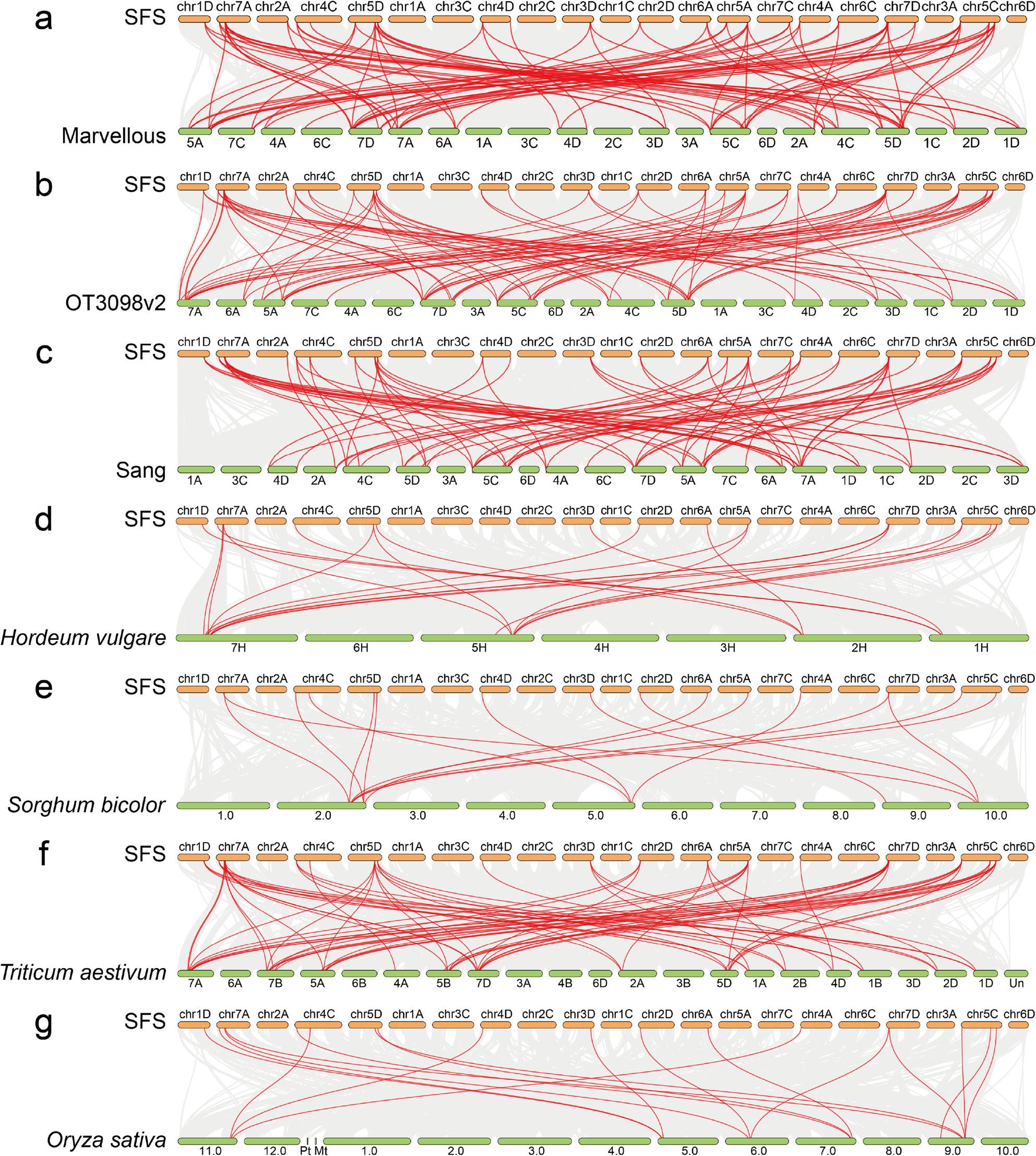 Figure 4
Figure 4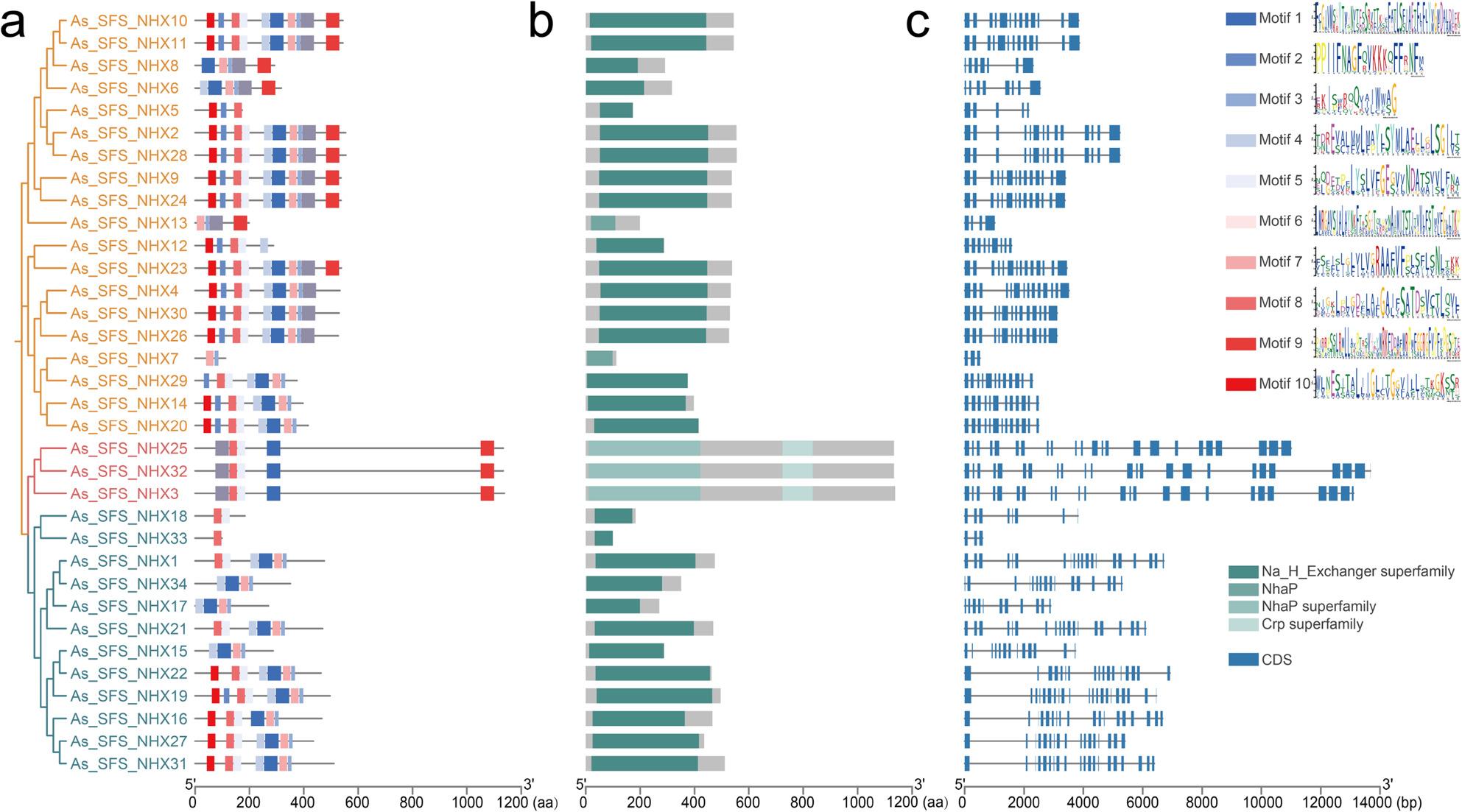 Figure 5
Figure 5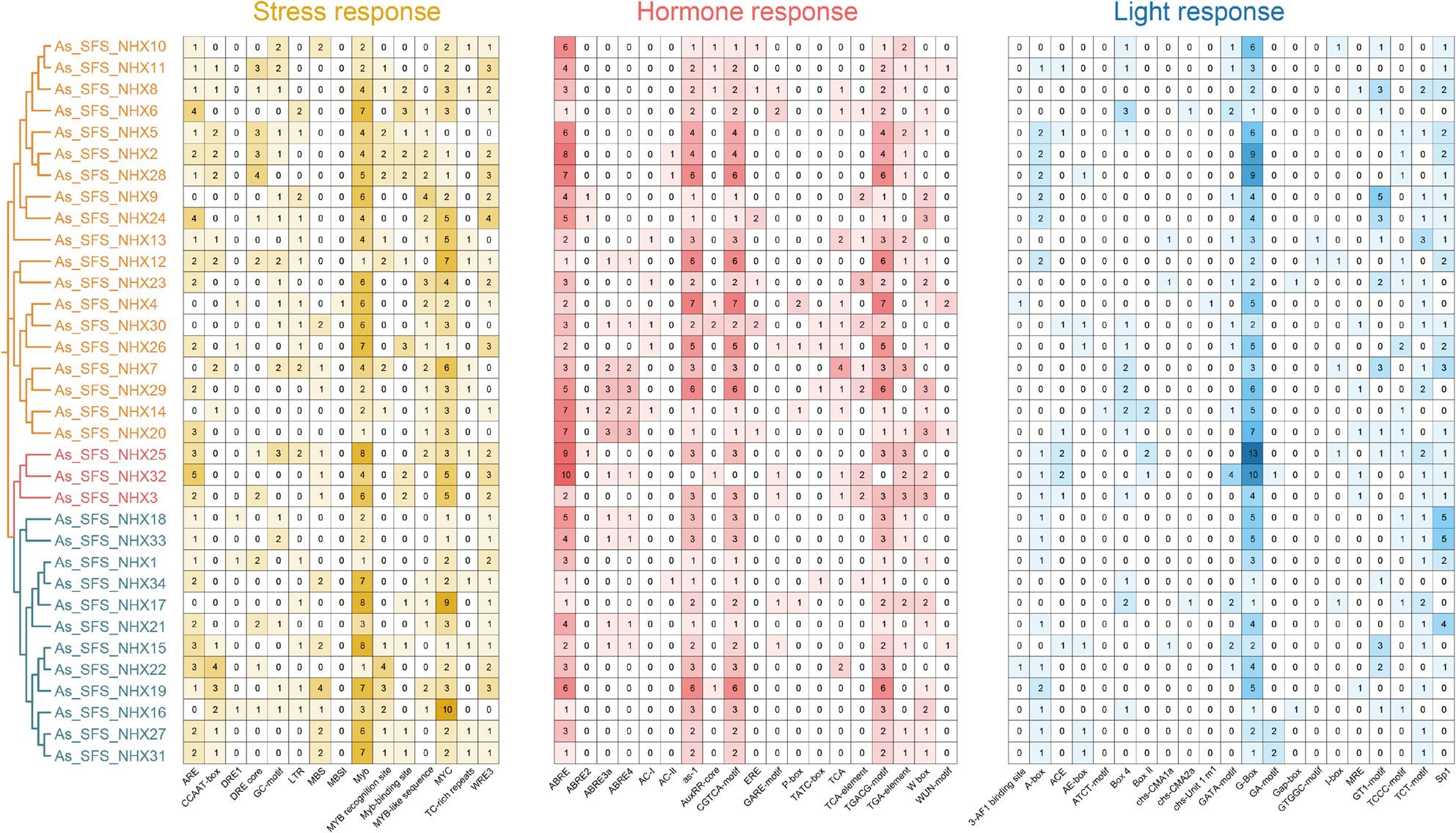 Figure 6
Figure 6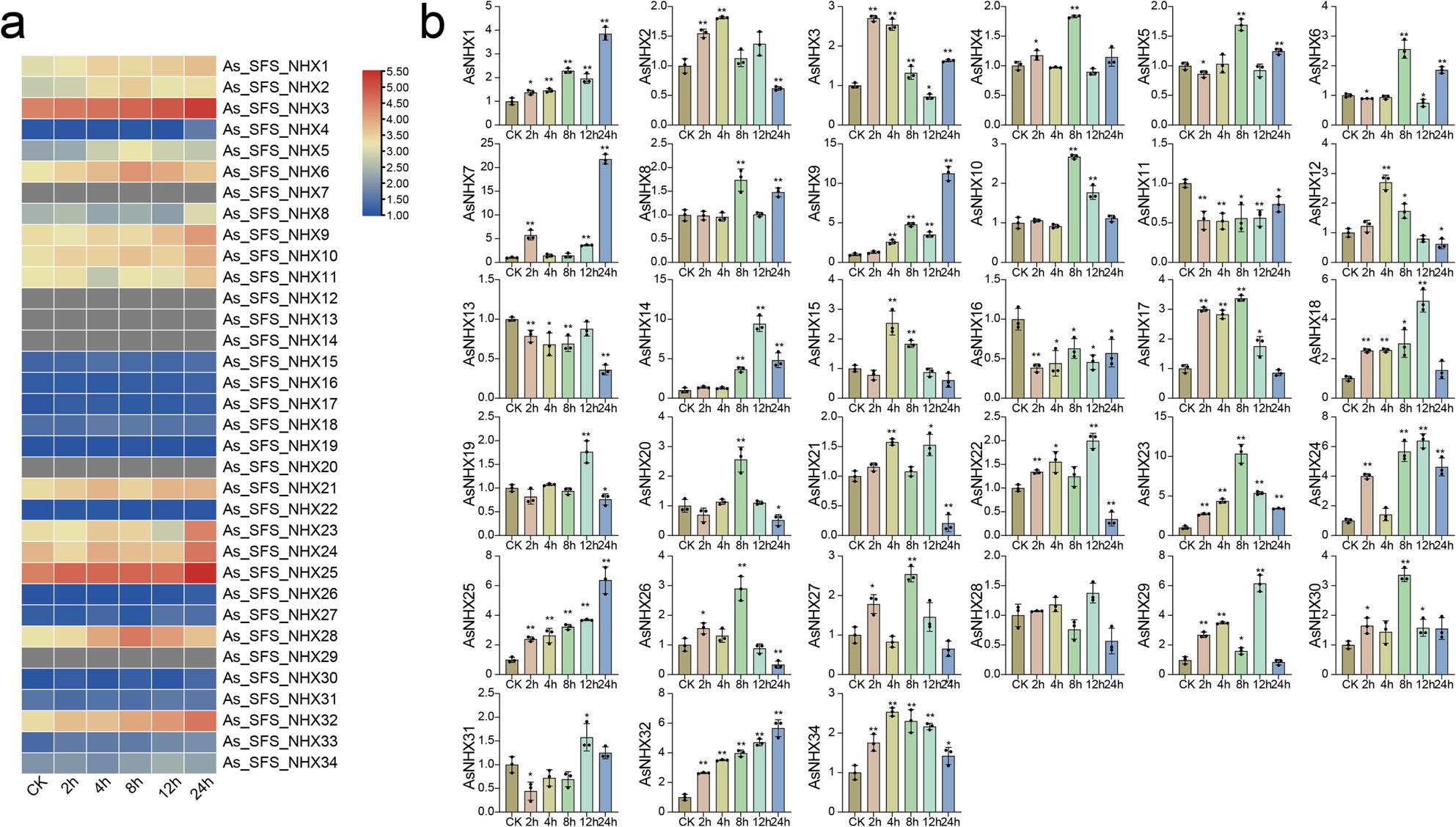 Figure 7
Figure 7 Figure 8
Figure 8- —National Key Research and Development Program of China
- —Government-Industry-Academia-Research-Promotion-Application-Finance of Hohhot Innovation Consortium Project
- —Taishan Scholar Program of Shandong, the Research on Key Technologies for Multi-Dimensional Microbial Culture Support & Product Industrialization
- —Qingdao New Energy Shandong Laboratory of Key Projects Programs
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Stress Responses and Tolerance · Plant nutrient uptake and metabolism · Plant Molecular Biology Research
Background
Soil salinization is a major abiotic constraint that threatens global agriculture and sustainable crop production. Approximately 800 million hectares of farmland are affected by salinity, with saline-alkali soils in China alone representing ~ 20% of the cultivated area [1–3]. Excess sodium accumulation disrupts photosynthesis, nutrient balance, and cellular integrity, leading to sharp yield losses across crop species [4]. Climate change and poor irrigation practices further intensify soil salinization, underscoring the urgent need to develop salt-tolerant varieties [5].
Plants employ multiple strategies to mitigate salt stress, with ionic homeostasis being a central factor in tolerance [6]. The conserved SALT OVERLY SENSITIVE (SOS) signaling pathway regulates sodium extrusion and sequestration through calcium-mediated signaling [7, 8]. NHX antiporters act as critical executors of this pathway, mediating Na⁺/H⁺ exchange to restore ion balance [9]. NHXs function in coordination with SOS pathway components, particularly CBL-CIPK complexes, to regulate sodium compartmentalization and exclusion [10]. While other adaptive mechanisms, such as osmolyte synthesis (e.g., proline, glycine betaine) and antioxidant activity (SOD, CAT, POD), contribute to salt tolerance, these processes ultimately depend on the establishment of ionic equilibrium, highlighting the central role of NHX transporters [11].
As members of the CPA1 family of cation/proton antiporters, NHXs facilitate electroneutral exchange of protons with sodium or potassium ions across membranes [12]. Based on subcellular localization and function, they are classified into Vac-class, Endo-class, and PM-class transporters [13–15]. Functional studies have established their significance in salt tolerance. For example, heterologous expression of AtNHX1 enhanced vacuolar sodium sequestration and salinity tolerance in transgenic plants [16]. In cotton, GbNHX2 expression was strongly induced by salt stress, contributing to ion balance [17]. Similarly, overexpression of OsNHX1 enabled plants to withstand 100 mM NaCl stress through enhanced sodium compartmentalization [18]. Collectively, these findings demonstrate the utility of NHX genes as targets for engineering salinity resilience.
Oat (Avena sativa L.), particularly the naked oat cultivar ‘Sanfensan (SFS)’, is an important hexaploid cereal cultivated globally in temperate regions [19, 20]. Its nutritional richness—β-glucans, proteins, and essential lipids—combined with adaptability to marginal soils, makes it valuable for both food and feed [21, 22]. Oats can thrive where other cereals fail, positioning them as promising candidates for cultivation in salinity-affected regions [23, 24]. Despite their agronomic significance, the NHX gene family in oat remains poorly studied compared to Arabidopsis (8 members), rice (7 members), wheat (30 members), and cotton (23–24 members) [25–28]. Key questions concerning their genome-wide distribution, duplication, evolutionary trajectories, and stress-related functions remain largely unexplored.
To bridge this gap, we performed the first systematic analysis of the NHX gene family in four representative oat cultivars, including SFS. The objectives were to: (1) identify NHX genes genome-wide and characterize their physicochemical features; (2) assess evolutionary relationships and duplication events via phylogenetic and synteny analyses; (3) evaluate expression dynamics and cis-regulatory elements under salt stress; and (4) predict protein–protein interaction networks to reveal regulatory roles. This study provides a comprehensive resource for dissecting NHX-mediated salt adaptation in oat and identifies genetic targets to guide marker-assisted breeding for enhanced salinity tolerance in cereal crops.
Materials and methods
Plant materials and salt stress treatment
The naked oat (Avena sativa var. nuda) cultivar ‘Huazao-2’ was used in this study. Uniform seeds were germinated at 24 °C under a 16-hour light/8-hour dark photoperiod. Seedlings at the two-leaf stage were transferred to hydroponic culture with Hoagland’s nutrient solution. At the three-leaf stage, plants were exposed to salt stress by adding 100 mM NaCl to the nutrient solution. Leaf samples were collected at 0, 2, 4, 8, 12, and 24 h post-treatment, immediately frozen in liquid nitrogen, and stored at − 80 °C for RNA extraction. Each treatment included three biological replicates, with three seedlings pooled per replicate.
Identification of the AsNHX gene family
Genomic data for four oat cultivars— ‘Marvellous’, ‘OT3098v2’, ‘Sang’, and ‘Sanfensan (SFS)’—were obtained from the OatBioDB database (http://waoOat.cn/). ‘Marvellous’, ‘OT3098v2’, and ‘Sang’ are hulled oat cultivars, while ‘SFS’ is a naked oat. Genomic data for Oryza sativa, Triticum aestivum, Arabidopsis thaliana, Sorghum bicolor, and Hordeum vulgare were retrieved from Ensembl Plants (http://plants.ensembl.org/index.html). The Hidden Markov Model (HMM) profile for the NHX domain (PF00999) was downloaded from Pfam (https://pfam.xfam.org) and searched against oat genomes using HMMER 3.0 [29] with an E-value cutoff of 1e^− 5^. In parallel, BLAST searches were conducted in TBtools [30] using known NHX protein sequences from rice, wheat, Arabidopsis, sorghum, and barley. Candidates identified by both approaches (E-value < 1e^− 5^) were retained as putative NHX genes. All candidates were further validated using the Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/cdd/). TBtools was used to calculate physicochemical properties of AsNHX proteins, including molecular weight and isoelectric point.
Phylogenetic, gene Structure, conserved Domain, and motif analyses
NHX protein sequences from the four oat cultivars and five reference species were aligned using ClustalW, and a phylogenetic tree was constructed in MEGA 7.0 with the Maximum Likelihood method and 1,000 bootstrap replicates [31]. The tree was visualized in iTOL [32]. Gene structures were determined using genome annotations. Conserved motifs were predicted with MEME (https://meme-suite.org/meme/) [33], and conserved domains were confirmed using the NCBI CDD database. (https://www.ncbi.nlm.nih.gov/cdd/). All results were visualized using TBtools [30]. Motif enrichment analysis was conducted using the Analysis of Motif Enrichment (AME) tool from MEME Suite v5.5.8. Statistical significance was evaluated with Fisher’s exact test, followed by Benjamini-Hochberg correction for multiple testing. Motifs with adjusted P-values < 0.05 were considered significantly enriched.
Cis-Regulatory element analysis
For each AsNHX gene, a 2.0-kb sequence upstream of the translation initiation codon (ATG) was defined as the promoter region. Cis-acting regulatory elements were predicted using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). Identified CREs were classified into functional categories (e.g., stress-responsive, hormone-responsive, light-responsive) and visualized as clustered heatmaps in TBtools [30].
Chromosomal localization and synteny analysis
The chromosomal positions of AsNHX genes were determined with TBtools using genome annotation files [30]. Synteny analysis was performed with MCScanX [34] to examine collinearity within the four oat genomes and between ‘SFS’ and other oat cultivars (‘Marvellous’, ‘OT3098v2’, ‘Sang’) as well as rice, wheat, sorghum, and barley. Ks (synonymous), Ka (nonsynonymous), and Ka/Ks ratios were calculated in TBtools for all homologous gene pairs [30].
Protein-Protein interaction (PPI) analysis
The PPI network of AsNHX proteins in ‘SFS’ was predicted using STRING (https://cn.string-db.org/), with Arabidopsis proteins as the reference model [35]. The network was visualized in Cytoscape 3.10.0 [36].
Transcriptome data analysis
Transcriptome datasets related to salt stress in oat were obtained from the NCBI Sequence Read Archive (BioProject accession PRJNA355375) (https://www.ncbi.nlm.nih.gov/sra/?term=Oat). The oat variety ‘HanYou-5’ was grown in Hoagland nutrient solution until the three-leaf stage and subsequently treated with 100 mM NaCl. Plant samples were collected at 2, 4, 8, 12, and 24 h post-treatment for transcriptome sequencing. Expression levels of NHX genes were quantified as log₂(TPM + 1), and expression patterns were visualized as heatmaps using TBtools [30].
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted using the RNAprep Pure Plant Kit (Tiangen, Beijing, China). First-strand cDNA was synthesized with the PrimeScript RT Reagent Kit (TaKaRa, Dalian, China). Gene-specific primers are listed in Table S7. AsACTIN was used as the internal control. qRT-PCR reactions were performed with the SYBR Green Pro Taq HS kit following the manufacturer’s protocol, using a Roche LightCycler 96 instrument (Roche, Basel, Switzerland). Amplification was conducted with a three-step PCR cycling program. Three biological replicates were analyzed for each sample. Relative expression levels were calculated using the 2^⁻ΔΔCT^ method [37]. Statistical significance was assessed using Student’s t-test.
Subcellular localization
NHX-eGFP was introduced into Agrobacterium GV3101 and grown overnight at 28 °C in LB medium. Cells were collected and resuspended in infiltration buffer (10 mM MgCl_2_; 10 mM MES-KOH, pH 5.5; 100 µM acetosyringone) to OD_600_ = 0.8, then pressure-infiltrated into leaves of 4-week-old Nicotiana benthamiana plants. After 24 h darkness, plants were transferred to standard conditions (16 h light/8 h dark) for 48 h, GFP was visualized using an Olympus FV3000 confocal microscope. Primer sequences are listed in Table S7.
Results
Identification, Phylogeny, and physicochemical properties of NHX genes
Using HMMER and BLASTP, 31, 30, 31, and 34 NHX family genes were identified in the oat cultivars ‘Marvellous’, ‘OT3098’, ‘Sang’, and ‘SFS’, respectively (Fig. 1a). The NHX proteins were systematically named according to chromosomal positions (Marvellous: As_Marvel_NHX1-31; OT3098: As_OT3098_NHX1-30; Sang: As_Sang_NHX1-31; SFS: As_SFS_NHX1-34) (Table S1). Physicochemical analysis revealed wide variation in sequence length, molecular weight, and isoelectric point (pI). In ‘SFS’, proteins ranged from 99 to 1138 aa, with pI values from 4.91 (As_SFS_NHX17) to 10.51 (As_SFS_NHX13), and molecular weights of 10.92 kDa (As_SFS_NHX33) to 126.29 kDa (As_SFS_NHX3). ‘Sang’ proteins spanned 153–1137 aa, with pI values of 5.13 (As_Sang_NHX15) to 9.07 (As_Sang_NHX26), and molecular weights of 16.55 kDa (As_Sang_NHX30) to 126.10 kDa (As_Sang_NHX2). In ‘OT3098’, proteins were 204–1137 aa, with pI values from 4.71 (As_OT3098_NHX10) to 9.13 (As_OT3098_NHX3), and molecular weights between 22.16 kDa (As_OT3098_NHX29) and 126.07 kDa (As_OT3098_NHX30). ‘Marvellous’ proteins ranged from 121–1137 aa, with pI values from 5.13 (As_Marvel_NHX15) to 8.69 (As_Marvel_NHX1), and molecular weights of 13.25 kDa (As_Marvel_NHX30) to 126.08 kDa (As_Marvel_NHX31). These findings highlight marked diversity in NHX family members across cultivars (Table S2).
Fig. 1. Identification and phylogenetic analysis of NHX genes in four oat genomes. a Genomic distribution of NHX genes across four oat genome assemblies. b Core and dispensable orthogroups (OGGs). The x-axis indicates the number of genomes in which an OGG is present. OGGs shared by all four genomes are designated as core, while those present in three, two, or one genome(s) are classified as dispensable. c Distribution of core and dispensable NHX genes among the four genomes. d Phylogenetic relationships of NHX proteins from four oat cultivars (Avena sativa, As), Oryza sativa (Os), Arabidopsis thaliana (At), Triticum aestivum (Ta), Hordeum vulgare (Hv), and Sorghum bicolor (Sb). Proteins cluster into three distinct groups, each highlighted in a different color
To assess conservation in the oat pan-genome, NHX genes were clustered into orthologous gene groups (OGGs) using a 95% amino acid similarity cutoff. The 126 identified genes formed 33 OGGs (Fig. 1b, Table S3). This total exceeded that of any individual genome, reflecting presence/absence variations (PAVs). Of the 33 OGGs, 10 were core NHX genes present in all cultivars, while 23 were dispensable (absent in at least one). The dispensable set included 2 OGGs shared by three cultivars, 5 shared by two, and 16 unique to single cultivars. ‘SFS’ contained the most core genes (25), whereas ‘OT3098’ had the fewest (19). In contrast, ‘OT3098’ carried the largest number of dispensable genes (11), while ‘Marvellous’ and ‘SFS’ each contained only 9 (Fig. 1c).
A phylogenetic tree of 181 NHX proteins from rice (Oryza sativa), wheat (Triticum aestivum), Arabidopsis (Arabidopsis thaliana), sorghum (Sorghum bicolor), barley (Hordeum vulgare), and the four oat cultivars confirmed close evolutionary relationships between oat and wheat NHX genes (Fig. 1d). Consistent with findings in Arabidopsis, the sequences grouped into three classes: vacuolar (Vac-class), predicted to localize to vacuoles; endosomal (Endo-class), localized to endosomal compartments; and plasma membrane (PM-class), associated with the plasma membrane. The Vac-class was the largest, comprising 70 oat proteins, followed by the Endo-class (37 proteins) and the PM-class (19 proteins).
Chromosomal localization and collinearity analysis of AsNHX genes
Chromosomal mapping revealed broad but uneven distribution of AsNHX genes (Fig. 2). In ‘Marvellous’, 31 genes were located on 15 chromosomes, with 10, 8, and 13 genes in the A, C, and D subgenomes, respectively (Fig. 2a). ‘OT3098’ carried 30 genes across 12 chromosomes (7, 7, and 16 in A, C, and D subgenomes) (Fig. 2b). In ‘SFS’, 34 genes were distributed across 16 chromosomes, with 10, 8, and 14 in the A, C, and D subgenomes, plus 2 on unplaced scaffolds (Un) (Fig. 2c). ‘Sang’ contained 31 genes on 15 chromosomes, with 9, 8, and 12 in the A, C, and D subgenomes, plus 2 on Un (Fig. 2d). Overall, AsNHX genes were enriched in the D subgenome. Notably, chromosomes 1 C, 2C, 3 A, 3 C, and 6D lacked NHX genes in all four cultivars.
Fig. 2. Chromosomal distribution of the NHX gene family in ‘Marvellous’ (a), ‘OT3098’ (b), ‘Sang’ (c), and ‘SFS’ (d). Yellow labels (left) indicate chromosome numbers, and red labels (right) indicate gene IDs
Gene duplication analysis identified 36, 34, 30, and 34 homologous gene pairs in ‘Marvellous’, ‘OT3098’, ‘Sang’, and ‘SFS’, respectively (Fig. 3). Evolutionary pressures were assessed using Ka/Ks ratios (Ka/Ks > 1: positive selection; =1: neutral; <1: purifying selection) (Table S4). Eight gene pairs—As_Marvel_NHX11/27, As_Marvel_NHX17/27, As_OT3098_NHX9/24, As_OT3098_NHX15/24, As_Sang_NHX11/25, As_Sang_NHX17/25, As_SFS_NHX14/29, and As_SFS_NHX20/29—displayed Ka/Ks > 1, indicating adaptive evolution. All others had Ka/Ks between 0 and 1, consistent with purifying selection.
Fig. 3. Collinearity of NHX genes in ‘Marvellous’ (a), ‘OT3098’ (b), ‘Sang’ (c), and ‘SFS’ (d). Syntenic gene pairs are connected by red lines
Collinearity analysis of naked oat ‘SFS’ with ‘Marvellous’, ‘OT3098’, ‘Sang’, Hordeum vulgare, Sorghum bicolor, Triticum aestivum, and Oryza sativa revealed extensive synteny (Fig. 4). ‘SFS’ shared 87 collinear pairs with ‘Marvellous’ (Fig. 4a), 86 with ‘OT3098’ (Fig. 4b), and 74 with ‘Sang’ (Fig. 4c). Cross-species comparison identified 22 collinear pairs with Hordeum vulgare (Fig. 4d), 16 with Sorghum bicolor (Fig. 4e), and 18 with Oryza sativa (Fig. 4g). The strongest homology was observed with Triticum aestivum, with 69 collinear pairs (Fig. 4f), consistent with phylogenetic clustering. These results suggest that orthologous genes, frequently shaped by duplication events, have contributed to oat evolution and expansion of the AsNHX family.
Fig. 4. Comparative collinearity of NHX genes between naked oat ‘SFS’ and ‘Marvellous’ (a), ‘OT3098’ (b), ‘Sang’ (c), Hordeum vulgare (d), Sorghum bicolor (e), Triticum aestivum (f), and Oryza sativa (g). Gray lines indicate all collinear blocks; red lines highlight collinear NHX gene pairs
Genome-Wide phylogenetic assessment of oat cultivars
To clarify phylogenetic relationships among the four oat cultivars, we performed a genome-wide species tree analysis using OrthoFinder. Contrary to initial inferences based on synteny pair counts, the species tree revealed a distinct topology: (SFS, (Sang, (Marvellous, OT3098))) (Fig. S1). This indicates that ‘SFS’ represents the earliest diverging lineage, followed by ‘Sang’, with ‘Marvellous’ and ‘OT3098’ forming the most closely related clade (bootstrap support = 0.70). These genome-wide phylogenetic results confirm the greater evolutionary divergence of naked oat ‘SFS’ from the hulled cultivars ‘Sang’, ‘Marvellous’, and ‘OT3098’.
Analysis of conserved Motifs, Domains, and gene structure of NHX genes
To explore the structural diversity of NHX genes in ‘SFS’, conserved motifs were analyzed using MEME, identifying 10 distinct motifs across their protein sequences (Fig. 5a). The sequence logos and detailed consensus sequences for each motif are provided in Fig. 5a and Table S5. PM-class NHX genes contained motifs 1, 5, 6, 8, and 9. Most Endo-class members carried motifs 1, 3, 4, 5, 7, and 8, while As_SFS_NHX33 contained only motif 8. Vac-class genes encompassed all 10 motifs. While motif composition and number varied significantly between classes, they were highly conserved within each class, supporting the classification system and indicating structural conservation among NHX proteins. The strict conservation of specific motif combinations within each class strongly supports the phylogenetic classification and suggests divergent functional specialization among NHX subfamilies.
Fig. 5. Motifs, conserved domains, and gene structures of NHX genes in ‘SFS’. a Top 10 conserved motifs of NHX proteins. The sequence logo for each motif is adjacent to the map. b Distribution of conserved domains. c Gene structures, with exons in blue boxes and introns in gray lines. Scale bars in (a) and (b) indicate amino acid length, while the scale in (c) represents nucleotide sequence length. Proteins are ordered according to a maximum-likelihood phylogenetic tree, with subfamilies color-coded as in Fig. 1
Motif enrichment analysis using AME quantified the relative abundance and significance of identified motifs. Motifs 7 and 3 were the most significantly enriched, present in 79.41% of NHX sequences but only 0.02% of whole-genome sequences, representing an approximate 4,000-fold enrichment (Table S6). Their conservation across the majority of NHX members indicates that these motifs encode critical functional domains potentially involved in transmembrane transport, ion binding, or protein–protein interactions typical of NHX exchangers. Notably, the combinatorial presence of multiple enriched motifs within individual NHX sequences suggests a modular architecture, where conserved regions may cooperate to mediate diverse functional roles during plant development and stress responses.
Domain analysis showed that, in addition to the Na_H_Exchanger superfamily domain, As_SFS_NHX13 and As_SFS_NHX7 harbored the NhaP domain, implicating them in K⁺/H⁺ transmembrane exchange (Fig. 5b). Moreover, PM-class members contained both NhaP and Crp superfamily domains. The Crp family is known to sense intracellular cAMP levels and contributes to salt stress responses. These results suggest that PM-class members play central roles in maintaining ion homeostasis and regulating salt stress adaptation in naked oat.
Gene structure analysis revealed marked variability in exon–intron organization (Fig. 5c). Intron numbers ranged from 2 to 23. PM-class genes NHX3, NHX25, and NHX32 contained the most introns (22–23). The Endo-class averaged 14.25 introns per gene, whereas the Vac-class carried the fewest, averaging 10.11 introns. Differences in intron number among the three classes likely contribute to gene length variation and further support structural conservation within oat NHX genes.
Analysis of Cis-Acting elements in NHX promoter regions
To investigate regulatory mechanisms underlying NHX expression, cis-acting elements were identified in the 2000 bp upstream promoter regions of the 34 NHX genes in ‘SFS’ using PlantCARE. These elements were grouped into three categories: stress-responsive (15 types), hormone-responsive (19 types), and light-responsive (21 types) (Fig. 6). A total of 577 stress-related elements were detected, associated with responses to low temperature, drought, anaerobic induction, and wounding. Hormone-related elements totaled 598, covering ABA, salicylic acid (SA), auxin (IAA), gibberellin (GA), and jasmonic acid (JA) signaling pathways. Among these, ABA-responsive elements (ABRE) were the most abundant, present in all genes, underscoring their central role in regulation. As_SFS_NHX32 contained the highest number of hormone-responsive elements (10), suggesting a strong involvement in hormone signaling. MeJA-responsive elements were widespread, occurring in all genes except As_SFS_NHX32.
Fig. 6Cis-acting regulatory elements in the promoters of 34 NHX genes in ‘SFS’. Elements are grouped into stress-responsive, hormone-responsive, and light-responsive categories. The heatmap depicts the distribution and frequency of elements, with numbers indicating their abundance per gene
All NHX genes also contained light-responsive elements, with the G-box being the most prevalent and present in every promoter. Notably, PM-class genes As_SFS_NHX3, As_SFS_NHX25, and As_SFS_NHX32 harbored the highest densities of stress-, hormone-, and light-responsive elements, indicating their importance in growth, development, and abiotic stress adaptation in naked oat.
Expression profiling of oat NHX genes under salt stress
Given the well-established role of NHX genes in salt tolerance, the expression of 34 AsNHX genes was assessed in naked oat exposed to NaCl for 0, 2, 4, 8, 12, and 24 h (Fig. 7a). Heatmap analysis of log₂(TPM + 1) values showed that As_SFS_NHX7/12/13/14/20/29 were undetectable, while As_SFS_NHX4/15/16/17/18/19/22/26/27/30/31/33/34 exhibited no significant changes under salt stress. In contrast, PM-class genes As_SFS_NHX3/25/32 responded strongly, with expression levels increasing steadily and peaking at 24 h, suggesting salt-stress regulation.
Fig. 7. Expression patterns of 34 NHX genes under salt stress. a Transcriptome profiling (RNA-seq) of 34 AsNHX genes in leaf under 100 mM NaCl treatment at 0 h (CK), 2 h, 4 h, 8 h, 12 h, and 24 h. The heatmap illustrates log₂(TPM + 1) values. The color scale in both heatmaps represents expression levels, from low (blue) to high (red). b qRT-PCR validation of AsNHX gene expression in leaf under identical salt stress conditions. Expression levels are presented relative to the 0 h control. Statistical significance between each treatment time point and the control was determined by Student’s t-test (*P < 0.05, **P < 0.01)
To validate transcriptome findings, naked oat seedlings were treated with 100 mM NaCl, and expression of 33 NHX genes was analyzed by qRT-PCR at the same time points (Fig. 7b). Expression of AsNHX33 could not be accurately quantified due to sequence identity with part of AsNHX18. The analysis revealed diverse expression patterns. AsNHX11 and AsNHX16 were significantly downregulated, while most genes were upregulated under salt treatment. Notably, AsNHX1/7/9/14/23/24/25/32 exhibited the strongest induction. Several genes displayed time-dependent dynamics, being either early- or late-responsive. For example, AsNHX3/12/15/17/18/23/30/34 were upregulated initially but declined later, whereas AsNHX31 decreased first and then increased. These patterns indicate that AsNHX genes contribute differentially to salt stress responses, with some functioning in early signaling and others in later adaptive processes.
Protein-Protein interaction network analysis of NHX genes
To identify potential partners of NHX proteins in salt stress adaptation, a protein–protein interaction (PPI) network was constructed using the STRING database (Fig. 8). No direct interactions among AsNHX proteins were predicted. Of the 34 AsNHX proteins, 17 were included in the PPI network, interacting primarily with members of the HKT and CBL families. HKT proteins mediate Na⁺ and K⁺ transport, while CBL proteins regulate intracellular Na⁺ and K⁺ homeostasis, enhancing salt tolerance. These associations suggest that NHX proteins act coordinately with other salt-responsive proteins to mediate adaptation. PM-class genes As_SFS_NHX3, As_SFS_NHX25, and As_SFS_NHX32 displayed more extensive interactions with HKT and CBL proteins, further underscoring their key role in ion regulation and salt stress response.
Fig. 8. Predicted protein-protein interactions (PPIs) of NHX proteins in the ‘SFS’ genome. Gray lines denote potential interactions; node size reflects the number of interacting partners
To validate potential interactions predicted by the PPI network, we analyzed the expression of interaction partners under the same salt stress conditions (Fig. S2). Genes including HKT1, HKT2, HKT9, and CBL10 were significantly upregulated, with induction kinetics overlapping those of core AsNHX genes (AsNHX3, AsNHX25), supporting potential functional cooperation in the salt stress response.
Subcellular localization of PM-class AsNHX proteins
To experimentally confirm plasma membrane localization of key AsNHX transporters, AsNHX3-GFP, AsNHX25-GFP, and AsNHX32-GFP constructs were transiently expressed in Nicotiana benthamiana leaves. Confocal imaging revealed fluorescence exclusively at the cell periphery, delineating the plasma membrane (Fig. S3). These results provide direct experimental evidence that AsNHX3, AsNHX25, and AsNHX32 are plasma membrane-localized, confirming their bioinformatic classification and supporting their role in Na^+^ exclusion to mitigate salt stress.
Discussion
Na⁺/H⁺ antiporters encoded by the NHX gene family are critical for plant salt tolerance by maintaining intracellular pH and ion homeostasis [15]. Our genome-wide analysis identified 126 NHX genes across four oat cultivars, revealing both conserved features and lineage-specific innovations. Variation in gene number (30–34) and the predominance of accessory OGGs highlight substantial PAV, potentially underlying oats’ adaptability to marginal soils. Gene family expansion through duplication is a common evolutionary strategy; for instance, in allopolyploid cotton, tetraploids possess NHX numbers nearly equivalent to the sum of their diploid progenitors [26]. Similar NHX expansions occur in soybean and alfalfa, indicating that duplication is a widespread mechanism to cope with environmental stress [38, 39].
Phylogenetic classification grouped oat NHX proteins into vacuolar (Vac), endosomal (Endo), and plasma membrane (PM) classes, consistent with classifications in Arabidopsis thaliana, rice, Populus trichocarpa, Beta vulgaris, kiwifruit, and tea plant [27, 28, 40–43]. This conserved classification suggests that NHX functional specialization emerged early in angiosperm evolution and remains highly conserved. The Vac-class was the largest, a trend observed in Beta vulgaris and Populus trichocarpa [17, 42], underscoring the fundamental role of vacuolar sodium sequestration. Strong collinearity and sequence similarity between oat and wheat NHX genes support a close evolutionary relationship and parallel mechanisms of gene duplication and diversification [27].
Our Ka/Ks analysis further illuminates the evolutionary pressures shaping this family. Consistent with findings in cotton, tea plants, and other species, most oat NHX homologous pairs were under purifying selection (Ka/Ks < 1), indicating constraint on their core functions [26]. However, several pairs (e.g., As_Marvel_NHX11/27) exhibited Ka/Ks > 1, a hallmark of positive selection often associated with adaptive evolution. These observations align with reports of stress-responsive NHX genes in cotton and wheat, suggesting ongoing adaptive evolutionary pressures [26, 44]. Subgenomic mapping revealed asymmetric NHX distribution, with the D subgenome contributing most genes, a phenomenon also reported in other polyploids and potentially linked to subgenome dominance in stress adaptation [45–47]. The phylogenetic position of ‘SFS’ as the earliest-diverging lineage may reflect its adaptation to specific environments. As a naked oat, SFS’s distinct evolutionary history could reflect genetic changes associated with hull loss, a trait emerging early during cultivar diversification.
Structural and motif analyses revealed class-specific organization and functional divergence. Ten conserved motifs showed distinct patterns across classes: PM-class genes consistently contained motifs 1, 5, 6, 8, and 9 and carried the Crp and NhaP superfamily domains, which are important for cAMP sensing, K⁺/H⁺ exchange, and salt stress response [48–50]. Vac-class members contained all 10 motifs, reflecting higher functional complexity. The universal presence of the amiloride-binding site is a conserved feature critical for transport activity, previously documented in Arabidopsis, Zanthoxylum, and cotton NHX proteins [26, 51, 52]. Variability in intron number, particularly the elevated count in PM-class genes, represents a conserved architectural trait observed in Populus, soybean, and cotton [26, 38, 53]. This diversity may underlie regulatory flexibility and alternative splicing, broadening the functional repertoire of NHX genes under stress.
Previous studies suggest that cis-acting elements play a crucial role in regulating plant stress tolerance, growth, and development [54]. Promoter analysis uncovered a complex regulatory landscape. All AsNHX promoters carried elements for abiotic stress (e.g., cold, drought), hormone signaling (e.g., ABA, SA, JA), and light response. The universal presence of ABRE elements suggests ABA-dependent regulation, consistent with studies in rice, Arabidopsis, and Brassica napus [55, 56]. he pervasive presence of MeJA-responsive motifs indicates potential cross-talk between jasmonic acid signaling and salt tolerance, a mechanism increasingly documented in species such as cotton and tomato [26, 57, 58]. Notably, PM-class members (As_SFS_NHX3,* As_SFS_NHX25*,* As_SFS_NHX32*) were highly enriched in stress- and hormone-responsive elements, highlighting their role as regulatory hubs. This pattern suggests that these transporters may function as master regulators of oat salt stress responses, analogous to NHX1 in cucumber [44].
Protein–protein interaction predictions further supported the central roles of PM-class genes. Although no direct interactions were detected among AsNHX proteins, extensive associations were identified with HKT transporters and CBL calcium sensors, which regulate ion fluxes and downstream kinases [59, 60]. In Arabidopsis and Populus, SOS1/NHX7 interacts with CBL4/SOS3 and CIPK24/SOS2 to mediate Na⁺ extrusion, whereas Vac-class NHXs cooperate with H⁺-pumps, including H⁺-ATPase and H⁺-PPase, to facilitate vacuolar Na⁺ sequestration [15, 53]. Highly connected PM-class members (As_SFS_NHX3/25/32) may act as hubs integrating Na⁺/K⁺ balance with Ca²⁺ signaling, thereby coordinating salt tolerance.
Expression profiling confirmed that most AsNHX genes were upregulated under salt stress, mirroring patterns in wheat and B. napus [12, 56]. PM-class members, particularly AsNHX3/25/32, exhibited strong induction with peak expression at 24 h post-treatment. In Arabidopsis, NHX7/SOS1 exports Na⁺ to maintain cytosolic ion balance and a favorable Na⁺/K⁺ ratio, enhancing salt tolerance [25]. Based on promoter enrichment and predicted interactions, we propose that AsNHX3/25/32 perform similar functions in oat. qRT-PCR validated significant upregulation of AsNHX1/7/9/14/23/24/25/32 under salt stress. Functional orthologs, such as TaNHX3 in wheat and OsNHX1 in rice, and GbNHX1/2 in cotton, have been shown to enhance salt tolerance in transgenic plants [18, 61]. Together, these findings identify strong candidates for functional studies and genetic improvement of oat salt tolerance.
In summary, this study systematically characterizes the NHX family in oat, revealing their evolutionary trajectories, structural divergence, cis-regulatory complexity, and transcriptional responses. PM-class members, in particular, emerge as central regulators of salt stress adaptation, coordinating ion transport and signaling pathways. These insights provide a theoretical basis for exploiting NHX genes in oat breeding and genetic improvement for salt tolerance.
Conclusions
We identified 126 NHX genes across four oat cultivars, classified into Vac-, Endo-, and PM-classes, with significant PAV and evidence of gene duplication as a major driver of family expansion. The D subgenome contributed most to gene retention, and several gene pairs exhibited positive selection. Motif and promoter analyses demonstrated subfamily-specific functional divergence, with PM-class members enriched for stress- and hormone-responsive elements and carrying key domains (Crp and NhaP). Interaction predictions suggest cooperation with HKT and CBL proteins in ion homeostasis, while expression profiling confirmed strong induction of AsNHX1/7/9/14/23/24/25/32 under salt stress. Collectively, these findings highlight the evolutionary diversification and functional specialization of the oat NHX family, providing a foundation for functional studies and genetic improvement of salt tolerance.
Supplementary Information
Supplementary Material 1. Table S1. List of 126 NHX gene family members identified in four oat genomes. Table S2. Physicochemical properties of 126 NHX genes. Table S3. Orthologous gene group (OGG) clustering of NHX genes across four oat genomes. Table S4. Ka, Ks, and Ka/Ks ratios for each NHX gene pair. Table S5. Basic information on AsNHX protein motifs.Table S6. Significantly enriched motifs in the NHX gene family. Table S7. Primers used in this study.
Supplementary Material 2. Fig. S1. Phylogenetic relationships among oat cultivars. Fig. S2. Expression patterns of potential NHX-interacting proteins under salt stress. The heatmap displays transcript levels (log₂(TPM + 1)) of genes encoding HKT and CBL family proteins, which were predicted to interact with AsNHX proteins, in leaf under 100 mM NaCl treatment at 0 h, 2 h, 4 h, 8 h, 12 h, and 24 h. Data are derived from the same RNA-seq dataset used in Figure 7a. Fig. S3. Subcellular localization of AsNHX3, AsNHX25, and AsNHX32 in Nicotiana benthamiana leaves.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Acharya BR, Zhao C, Reyes LAR, Ferreira JFS, Sandhu D. Understanding the salt overly sensitive pathway in Prunus: Identification and characterization of NHX, CIPK, and CBL genes. The Plant Genome. 2024;17:e 20371. 10.1002/tpg 2.20371.10.1002/tpg 2.20371 PMC 1280724737493242 · doi ↗ · pubmed ↗
- 2Sharma P, Mishra S, Pandey B, Singh G. Genome-wide identification and expression analysis of the NHX gene family under salt stress in wheat (Triticum aestivum L). Front Plant Sci. 2023;14. 10.3389/fpls.2023.1266699.10.3389/fpls.2023.1266699 PMC 1072605538111881 · doi ↗ · pubmed ↗
- 3Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v 10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;D 447–452. 10.1093/nar/gku 1003. 43 Database issue.10.1093/nar/gku 1003 PMC 438387425352553 · doi ↗ · pubmed ↗
- 4Haque MF, Tarusawa T, Ushida C, Ito S, Himeno H. c AMP-CRP-activated E. coli causes growth arrest under stress conditions. Front Microbiol. 2025;16. 10.3389/fmicb.2025.1597530.10.3389/fmicb.2025.1597530 PMC 1242613540950604 · doi ↗ · pubmed ↗