Mantle Cell Lymphoma with Persistent Massive Pleural Effusions Requiring Invasive Mechanical Ventilation and Bilateral Continuous Thoracic Drainage
Taichiro Tokura, Youhei Imai, Satoshi Sakai, Reina Saga, Hiroko Hidai, Sayuri Motomura

TL;DR
A rare case of mantle cell lymphoma caused severe lung fluid buildup requiring ventilation and chest drainage, but treatment led to full recovery.
Contribution
Reports a rare clinical case of MCL with massive pleural effusions requiring mechanical ventilation and bilateral drainage.
Findings
Pleural effusions subsided after immunochemotherapy, allowing extubation and removal of chest tubes.
Complete remission was achieved with resolution of lymphadenopathy and bone marrow infiltration.
Prompt diagnosis and treatment were critical for managing this rare MCL complication.
Abstract
Background and Clinical Significance: Mantle cell lymphoma (MCL) frequently involves bone marrow, gastrointestinal tract, and hepatosplenomegaly, whereas pleural effusions are uncommon. Cases requiring invasive mechanical ventilation and thoracic drainage are rare. We report a case of MCL with persistent massive pleural effusions requiring invasive mechanical ventilation and bilateral continuous thoracic drainage. Case Presentation: A 71-year-old woman presented with dyspnea and was found to have bilateral pleural effusions and generalized lymphadenopathy. Shortly after admission, she developed acute respiratory failure due to pleural effusions and required invasive mechanical ventilation. Right-sided continuous thoracic drainage was initiated. Thereafter, more than 1 L of pleural fluid was drained each day. Flow cytometry of the pleural fluid showed CD5-positive B cells with kappa…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
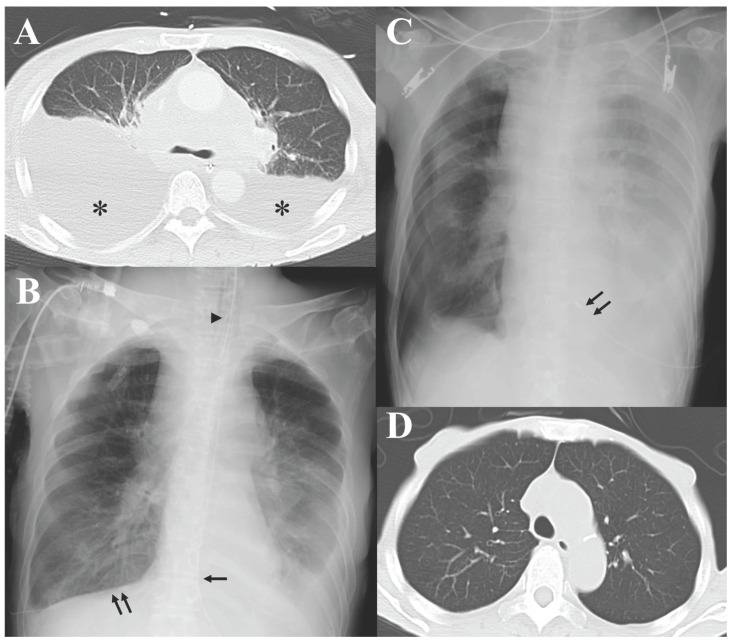 Figure 1
Figure 1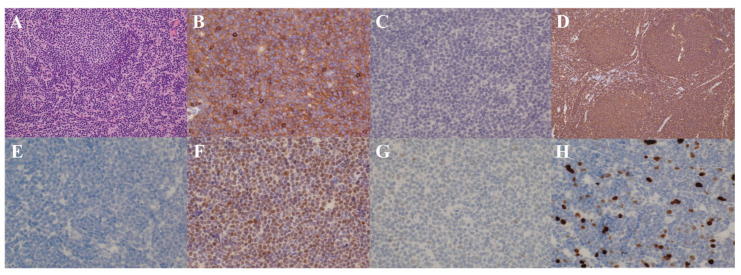 Figure 2
Figure 2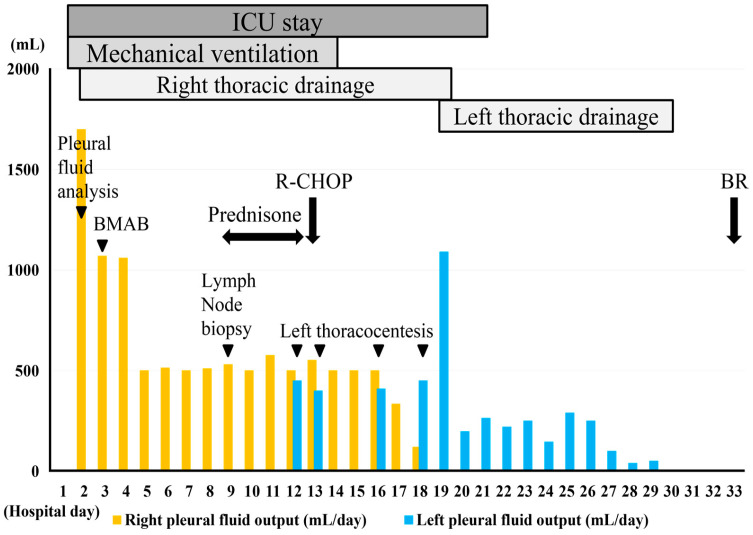 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLymphoma Diagnosis and Treatment · Lymphadenopathy Diagnosis and Analysis · CNS Lymphoma Diagnosis and Treatment
1. Introduction and Clinical Significance
Mantle cell lymphoma (MCL) is an uncommon mature B-cell neoplasm characterized by the proliferation of medium-sized lymphocytes [1,2]. The translocation t(11;14)(q13;q32) leading to constitutive overexpression of cyclin D1 is a pathognomonic feature [1,2]. Patients typically present with generalized lymphadenopathy and are classified as Ann Arbor/Lugano stage III or IV [3]. Extranodal involvement is observed in approximately 75–90% of patients, including infiltration of bone marrow (53–92%), gastrointestinal tract (88%), peripheral blood (34–50%), spleen (27%), and liver (8–25%) [4,5,6,7]. However, pleural involvement and pleural effusions are rare, and reports of these as initial presenting symptoms are even fewer [8].
Clinically, MCL exhibits heterogeneous behavior, ranging from aggressive disease to indolent forms that may not require treatment for years [4]. This heterogeneity is partly reflected by two major subtypes: the more common conventional MCL (cMCL) and the less frequent leukemic non-nodal MCL (nnMCL) [1,2]. Both subtypes are clearly distinct in molecular characteristics and clinical presentation. cMCL very frequently harbors complex karyotypes with several driver gene alterations (median 7 per case), simultaneously affecting multiple pathways [1]. It presents with generalized lymphadenopathy and follows a more aggressive course [9]. In contrast, nnMCL usually displays a simple karyotype, dominated by t(11;14), with fewer driver gene alterations (median <2 per case) [1,10]. It does not cause lymphadenopathy, frequently presents with lymphocytosis and splenomegaly, and generally exhibits an indolent course with superior outcomes [9].
Current treatment paradigms are not subtype-specific. For both subtypes, induction immunochemotherapy, such as R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) or BR (bendamustine and rituximab), is recommended for symptomatic stage III or IV [4]. In recent years, the addition of Bruton tyrosine kinase (BTK) inhibitors, such as ibrutinib and acalabrutinib, to R-CHOP or BR has further improved clinical outcomes [4]. In addition, in patients <65 years and otherwise fit, high-dose consolidation therapy followed by autologous hematopoietic stem cell transplantation is the current standard of treatment [4]. Across subtypes, pleural involvement and pleural effusions are rare, and the need for invasive mechanical ventilation or continuous thoracic drainage during treatment is extremely rare. In particular, there are very few cases of de novo MCL requiring these interventions before diagnosis.
Here, we report a case of de novo MCL requiring invasive mechanical ventilation and bilateral continuous thoracic drainage due to life-threatening respiratory failure caused by persistent massive pleural effusions.
2. Case Presentation
A 71-year-old woman with no significant past medical history presented to a local hospital in September 2025, complaining of dyspnea. Chest-abdominal computed tomography (CT) revealed bilateral pleural effusions and lymphadenopathy in the cervical, mediastinal, and para-aortic regions. On 16 September, she was referred to our hospital for further evaluation and was admitted the same day. Physical examination revealed palpable right cervical lymphadenopathy and decreased breath sounds over the right lung field. No lower extremity edema or other significant abnormal findings were noted. Her vital signs on admission were: alert consciousness, body temperature 36.6 °C, heart rate 84 bpm, blood pressure 131/59 mmHg, respiratory rate 18/min, and SpO_2_ 95% on room air. Her Eastern Cooperative Oncology Group (ECOG) performance status was 2. The electrocardiogram showed no remarkable abnormalities. Laboratory tests showed a white blood cell count of 9300/μL (neutrophils 59.5%, lymphocytes 9.0%, monocytes 5.0%, and abnormal lymphocytes with indented nuclei and a high nuclear-to-cytoplasmic ratio 26.5%), hemoglobin 14.3 g/dL, platelets 29.7 × 10^3^/μL, lactate dehydrogenase (LDH) 278 U/L, C-reactive protein (CRP) 0.15 mg/dL, N-terminal pro-B-type natriuretic peptide (NT-proBNP) 207 pg/mL, and soluble IL-2 receptor (sIL-2R) 3730 U/mL, and hepatitis B and C virus serologies were negative.
On the night of admission, she developed sudden respiratory failure, with her SpO_2_ dropping to 81%. Although supplemental oxygen was started, her oxygen saturation did not improve, so endotracheal intubation was performed, and invasive mechanical ventilation was initiated. Contrast-enhanced CT from the neck to the pelvis demonstrated lymphadenopathy in the bilateral cervical, supraclavicular, and hilar regions, as well as in the mediastinal, para-aortic, and mesenteric areas. Bilateral pleural effusions, more prominent on the right side, were also observed (Figure 1A). Transthoracic echocardiography showed preserved cardiac function with no remarkable abnormalities. Her acute respiratory failure was considered to be due to the pleural effusion, which worsened in the supine position. A chest tube was inserted into the right pleural cavity, and continuous thoracic drainage was initiated (Figure 1B). Although her respiratory status became stable, more than 1 L of pleural fluid continued to be drained each day thereafter.
Pleural fluid analysis showed a yellowish-brown appearance, a pH of 8.5, a nucleated cell count of 1109/µL with 34.5% abnormal lymphocytes, LDH 228 U/L, glucose 157 mg/dL, total protein 3.7 g/dL, albumin 2.6 g/dL, and a positive Rivalta test. Concurrent serum total protein and albumin levels were 5.9 g/dL and 3.5 g/dL, respectively, yielding a pleural fluid-to-serum protein ratio of 0.63, a serum-to-pleural fluid albumin gradient of 0.9 g/dL, and a serum-to-pleural fluid total protein gradient of 2.2 g/dL, consistent with an exudative effusion. Its culture was negative. Cytological examination revealed numerous large abnormal lymphocytes with irregular nuclei, classified as class V. Flow cytometry of the pleural fluid identified a cell population, slightly larger than the lymphocyte fraction, that was CD5^+^, CD19^+^, CD20^+^, and CD23^−^, with a κ/λ ratio of 23, suspicious for B-cell lymphoma. Bone marrow aspiration and biopsy revealed that abnormal lymphocytes with the same immunophenotype accounted for approximately 30% of the nucleated cells. On hospital day 9, an excisional biopsy of a right cervical lymph node was performed. As shown in Figure 2, histopathological examination of the lymph node demonstrated diffuse proliferation of medium-sized, monomorphic abnormal B lymphocytes with an immunophenotype of CD5^+^, CD10^−^, CD19^+^, CD20^+^, CD23^−^, BCL2^+^, BCL6^+^, cyclin D1^+^, SOX11^+^, p53^−^, and κ^+^, with a Ki-67 index of approximately 20%. Fluorescence in situ hybridization (FISH) for IgH/CCND1 on the lymph node specimen showed fusion signals in 94.0% of analyzed cells. Conventional karyotyping revealed a normal karyotype. The pleural fluid cell block and bone marrow specimens demonstrated similar findings. Taken together, these results established a diagnosis of MCL, stage IV. According to the MCL International Prognostic Index (MIPI), she was classified as high risk [11]. The Ki-67-based combined MIPI (MIPI-c) placed her as high-intermediate risk [12].
On hospital day 9, following the lymph node biopsy, prephase therapy with prednisone 100 mg was initiated (Figure 3). Subsequently, her respiratory status gradually improved, and she was weaned from mechanical ventilation. On hospital day 13, because only CD20-positive B-cell lymphoma was suspected and definitive histopathology was still pending, and because her critical condition left no time to await the final diagnosis, the R-CHOP regimen was initiated, consisting of vincristine 1.5 mg (1.4 mg/m^2^), doxorubicin 53 mg (50 mg/m^2^), and cyclophosphamide 785 mg (750 mg/m^2^), each administered at 70% of the standard dose, with rituximab 560 mg (375 mg/m^2^). After initiation of R-CHOP, the right pleural effusion decreased, allowing removal of the right chest tube. A left-sided pleural effusion then required continuous thoracic drainage for a short time (Figure 1C), but the drainage output gradually decreased, and the left chest tube was also removed. From the second cycle onward, after MCL diagnosis was confirmed, the R-CHOP regimen was switched to the BR regimen based on evidence from a randomized trial implying better 5-year progression-free survival with BR compared with R-CHOP [13]; the BR regimen consisted of bendamustine 96 mg (70% of the standard 90 mg/m^2^ dose) and rituximab 560 mg (375 mg/m^2^). Because her ECOG performance status had deteriorated to 3–4, we prioritized tolerability and avoidance of treatment-related toxicity; therefore, we did not add ibrutinib to BR and administered bendamustine at a reduced dose [4,14]. After removal of both chest tubes, the bilateral pleural effusions resolved, with no re-accumulation thereafter (Figure 1D). Furthermore, generalized lymphadenopathy regressed, and bone marrow examination revealed resolution of lymphoma infiltration, resulting in complete remission (CR).
3. Discussion
Our patient developed persistent massive pleural effusions requiring invasive mechanical ventilation and bilateral continuous thoracic drainage. Pleural effusion is an uncommon manifestation of MCL, and it is even more unusual for MCL to present with life-threatening respiratory failure requiring invasive mechanical ventilation and continuous thoracic drainage prior to diagnosis. Accordingly, we conducted a literature review of reported cases of MCL with pleural effusion.
On 5 January 2026, we searched PubMed using the following query: (“mantle cell lymphoma” OR “mantle-cell lymphoma” OR “MCL” OR “Lymphoma, Mantle-Cell” [MeSH]) AND (pleural OR effusion OR hydrothorax OR hemothorax OR chylothorax). This search yielded 132 articles. After reviewing all retrieved articles, we identified reports in which pleural effusion was attributable to MCL based on pleural fluid evaluation, and we summarized eligible articles in Table 1 [15,16,17,18,19,20,21,22,23]. Similarly to our case, pleural effusion most often developed before treatment, and bilateral involvement was the most frequently reported distribution. In terms of the pathophysiology of pleural effusion, all published cases described exudative effusions. Given MCL’s propensity to infiltrate extranodal sites, these effusions are most plausibly attributable to direct pleural infiltration by lymphoma. In our patient, the pleural effusion was also exudative, and MCL cells were identified in the pleural fluid by flow cytometry and on the cell block, supporting direct pleural infiltration as the mechanism of pleural fluid accumulation. Regarding supportive interventions, only one report by Masha et al. described the need for continuous thoracic drainage [19]. To our knowledge, no prior report has documented the use of invasive mechanical ventilation in this context. R-CHOP and BR were the most commonly selected treatment regimens. In the report by Anai et al., concomitant tuberculosis was present and was treated accordingly [22]. Outcomes were poor in the published cases. Of the nine reported patients, five died, and infectious complications accounted for some of these deaths. Death directly attributable to progressive MCL was described in two reports by Safa et al. and Keklik et al., in which R-CHOP failed to control the disease [17,20]. In contrast, although our patient required invasive mechanical ventilation and continuous thoracic drainage, immunochemotherapy rapidly reduced pleural output, leading to durable resolution of pleural effusions and CR. Notably, none of the nine reports provided details on the MCL subtype. Clinically, our patient presented with generalized lymphadenopathy and aggressive, life-threatening pleural effusions, which are more compatible with cMCL. However, cMCL frequently exhibits a complex karyotype, whereas our patient had a normal karyotype. Further characterization using comprehensive genomic profiling, such as whole-genome sequencing (WGS), may be required to clarify the underlying biology in such cases. Accumulation of additional cases with integrated genomic data will be important to better define the clinical course and prognosis of MCL complicated by pleural effusion.
In contrast, pleural effusion is relatively common in aggressive B-cell lymphomas such as diffuse large B-cell lymphoma (DLBCL). In some of these aggressive B-cell lymphomas, aberrant cyclin D1 expression due to CCND1 rearrangement can occur, making pathological differentiation from MCL challenging [1,24]. In such situations, CD5 and SOX11 are useful discriminatory markers; their co-expression strongly favors MCL over cyclin D1–positive aggressive B-cell lymphomas [2,24]. In our patient, the lymphoma cells were positive for cyclin D1, CD5, and SOX11, thereby firmly supporting the diagnosis of MCL rather than cyclin D1-positive DLBCL despite the aggressive clinical presentation with massive pleural effusions.
Several biological features may be relevant to the aggressive clinical behavior. BCL6-positive MCL has been reported to be associated with an unfavorable prognosis [25]. Our patient’s MCL was BCL6-positive, which may, at least in part, explain the aggressive clinical course with pleural effusion. In addition, TP53 aberrations, including TP53 mutations and deletions, are regarded as the most robust molecular prognosticators and are associated with treatment resistance and poor outcomes [26]. Although p53 immunohistochemistry was negative in our case, truncating TP53 variants may not be detected by immunohistochemistry [4]. Therefore, assessment by DNA sequencing would be preferable to more definitively evaluate TP53 status. Beyond these immunohistochemical markers, genomic complexity, defined as a complex karyotype on conventional cytogenetics or more than three copy-number variations (CNVs), has emerged as an independent poor prognostic marker in patients treated with either immunochemotherapy or BTK inhibitors, and these findings have been replicated using WGS [2,4]. Although WGS was not performed in our patient, the severity of presentation raises the possibility of underlying genomic complexity.
To date, no dedicated studies have addressed whether MCL with pleural effusion has a worse prognosis than MCL without pleural effusion. However, several reports indicate that malignant lymphoma presenting with pleural effusion at initial diagnosis is associated with an adverse prognosis [27]. In DLBCL complicated by serous effusions, including pleural effusion, the presence of malignant effusion has been identified as an independent poor prognostic factor in multivariate analysis [28]. Moreover, a baseline pleural effusion volume ≥200 mL has been reported to correlate with inferior survival [29]. In our patient, the malignant pleural effusion clearly exceeded this threshold at presentation, and thoracic drainage consistently yielded more than 1 L/day. Although MIPI and MIPI-c were used for risk stratification in MCL, neither index explicitly incorporates pleural effusions. Whether pleural effusion represents an independent adverse prognostic marker in MCL remains unknown because dedicated cohorts are lacking. Nevertheless, by cautious analogy to DLBCL, malignant pleural effusions may identify a biologically aggressive subset with higher tumor burden and extranodal spread in MCL. This prognostic reasoning directly influenced our clinical decision-making, such as early initiation of prephase prednisone and R-CHOP without waiting for final histopathology. Afterward, we switched to BR, which has been associated with more favorable 5-year progression-free survival [13]. However, in a younger and fitter patient, the addition of a BTK inhibitor, such as ibrutinib or acalabrutinib, to R-CHOP or BR would also be a reasonable therapeutic option [4,14].
Given the rarity of this aggressive clinical course and her unfavorable risk stratification by the MIPI (high risk) and the MIPI-c (high-intermediate risk), relapse remains a plausible concern. Nevertheless, treatment options for relapsed or refractory (R/R) MCL have advanced substantially in recent years. Anti-CD19 chimeric antigen receptor (CAR) T-cell therapy has emerged as a highly active salvage option for R/R MCL. In ZUMA-2 cohort 3, brexucabtagene autoleucel achieved a 91% overall response rate with a 73% CR rate, with favorable 12-month estimates for progression-free and overall survival [30]. Likewise, lisocabtagene maraleucel demonstrated durable remissions in the TRANSCEND NHL 001 MCL cohort [31]. In parallel, the CD20 × CD3 T-cell–engaging bispecific antibody glofitamab has also shown promising efficacy in R/R MCL [32]. If our patient were to relapse, these novel therapies could still offer clinically meaningful benefit and represent important salvage options.
Despite these aggressive features, our patient achieved CR. We attribute this favorable outcome to early initiation of prephase prednisone and prompt introduction of immunochemotherapy with R-CHOP, followed by BR after the diagnosis of MCL was confirmed. This treatment strategy effectively reduced pleural output, allowed discontinuation of mechanical ventilation and thoracic drainage, and ultimately led to CR. Therefore, our case illustrates that even in de novo MCL presenting with massive pleural effusions and respiratory failure, a combination of appropriate ventilatory management, timely thoracic drainage, and rapid initiation of immunochemotherapy can be both feasible and potentially lifesaving. This case was also shared in our regular institutional educational conference, reinforcing its educational value and emphasizing the importance of timely diagnosis with appropriate treatment in similar presentations.
Because large-scale data specifically focusing on MCL with pleural effusion are lacking, the long-term risk of relapse and overall prognosis in such cases remain uncertain. Accumulation of additional cases and larger cohort studies is needed to clarify the prognostic impact of pleural effusion in MCL and to optimize treatment strategies for this rare but clinically challenging presentation.
4. Conclusions
De novo MCL complicated by persistent massive pleural effusions requiring invasive mechanical ventilation and continuous thoracic drainage before diagnosis is rare. However, in critically ill patients with rapidly progressive and otherwise unexplained massive pleural effusions, malignant lymphoma should be considered early, and prompt diagnostic evaluation should be pursued. An integrated approach comprising a thorough diagnostic workup, appropriate ventilatory management, timely thoracic drainage, and prompt initiation of immunochemotherapy can reduce pleural output, enable extubation, and be lifesaving. Clinicians should recognize that MCL rarely presents with persistent massive pleural effusions.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1López C. Silkenstedt E. Dreyling M. BeàS. Biological and clinical determinants shaping heterogeneity in mantle cell lymphoma Blood Adv.202483652366410.1182/bloodadvances.202301176338748869 PMC 11284685 · doi ↗ · pubmed ↗
- 2Campo E. Jaffe E.S. Cook J.R. Quintanilla-Martinez L. Swerdlow S.H. Anderson K.C. Brousset P. Cerroni L. de Leval L. Dirnhofer S. The International Consensus Classification of Mature Lymphoid Neoplasms: A report from the Clinical Advisory Committee Blood 202214012291253 Erratum in Blood 2023, 141, 437. https://doi.org/10.1182/blood.202201901610.1182/blood.202201585135653592 PMC 9479027 · doi ↗ · pubmed ↗
- 3Silkenstedt E. Dreyling M. Mantle cell lymphoma-Update on molecular biology, prognostication and treatment approaches Hematol. Oncol.202341364210.1002/hon.314937294961 · doi ↗ · pubmed ↗
- 4Jerkeman M. Aurer I. Campo E. Cheah C.Y. Clark J. Doorduijn J. Eyre T.A. Fehr M. GinéE. Gomes da Silva M. EHA-EU MCL network guidelines for diagnosis and treatment of mantle cell lymphoma Hema Sphere 20259 e 7023310.1002/hem 3.7023341132246 PMC 12541557 · doi ↗ · pubmed ↗
- 5Romaguera J.E. Medeiros L.J. Hagemeister F.B. Fayad L.E. Rodriguez M.A. Pro B. Younes A. Mc Laughlin P. Goy A. Sarris A.H. Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma Cancer 200397586591 Erratum in Cancer 2003, 97, 313110.1002/cncr.1109612548600 · doi ↗ · pubmed ↗
- 6Argatoff L.H. Connors J.M. Klasa R.J. Horsman D.E. Gascoyne R.D. Mantle cell lymphoma: A clinicopathologic study of 80 cases Blood 1997892067207810.1182/blood.V 89.6.20679058729 · doi ↗ · pubmed ↗
- 7Baheti A.D. Tirumani S.H. Sewatkar R. Sachin S.S. Shinagare A.B. Ramaiya N.H. MDCT of extranodal mantle cell lymphoma: A single institute experience Abdom. Imaging 2015401693169910.1007/s 00261-015-0389-925724714 · doi ↗ · pubmed ↗
- 8Vega F. Padula A. Valbuena J.R. Stancu M. Jones D. Medeiros L.J. Lymphomas involving the pleura: A clinicopathologic study of 34 cases diagnosed by pleural biopsy Arch. Pathol. Lab. Med.20061301497150210.5858/2006-130-1497-LITPAC 17090191 · doi ↗ · pubmed ↗