Redox-Switchable Halogen Bonding in Haloanthracene Mediators Enables Efficient Electrocatalytic C–N Coupling
Atsuki Hirama, Kayo Suda, Shohei Yoshinaga, Moto Kikuchi, Su-Gi Chong, Azusa Kikuchi, Yusuke Ishigaki, Daisuke Yokogawa, Mahito Atobe, Naoki Shida

TL;DR
This paper introduces redox-switchable halogen bonding in haloanthracene mediators to improve electrocatalytic C–N bond formation.
Contribution
The novel use of redox-switchable halogen bonding to control reactivity and selectivity in electrocatalysis is presented.
Findings
Iodoanthracene derivative 1a showed superior catalytic performance in C–N coupling.
Mediator 1h, with a 3,5-bis(trifluoromethyl)phenyl group, achieved high yields in short reaction times.
Computational studies confirmed that halogen bonding in the radical cation state enhances N–H acidity and PCET.
Abstract
We report the development of redox mediators based on 9-halo-10-arylanthracenes that engage in halogen bonding only upon one-electron oxidation. This redox-switchable interaction enables an effective substrate preorganization and promotes intramolecular C–N bond formation via electrocatalysis. Systematic evaluation of halogenated mediators (1a–1c) across various N-protected 2-aminobiphenyl substrates revealed that the iodoanthracene derivative 1a exhibited superior catalytic performance. Building on this, we synthesized a series of 10-aryl-substituted iodoanthracenes (1d–1h) to further optimize the mediator structure. Kinetic analysis by foot-of-the-wave analysis identified 1h, bearing a 3,5-bis(trifluoromethyl)phenyl group, as a highly active mediator with an apparent rate constant over an order of magnitude higher than that of its counterparts. Bulk electrolysis experiments…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1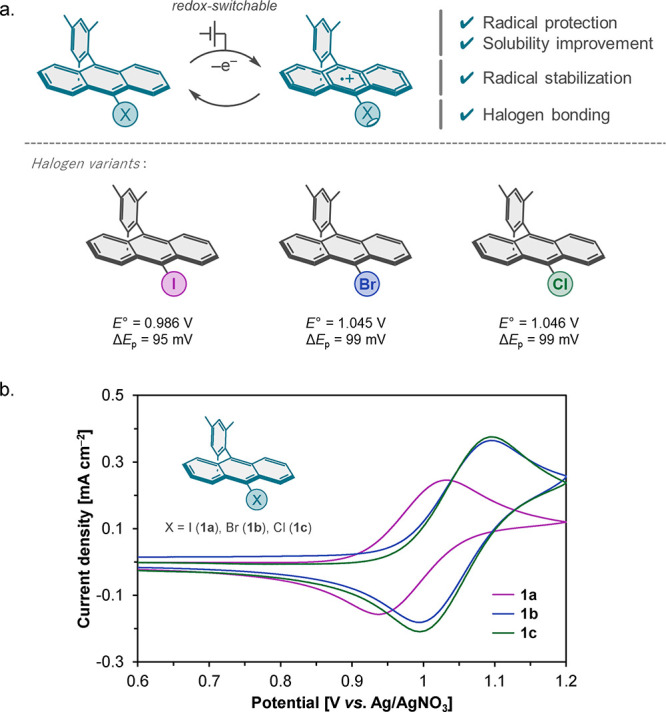 2
2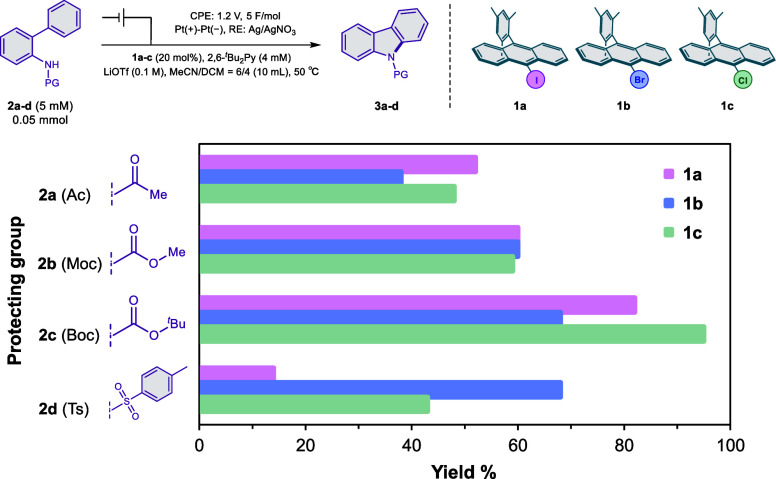 3
3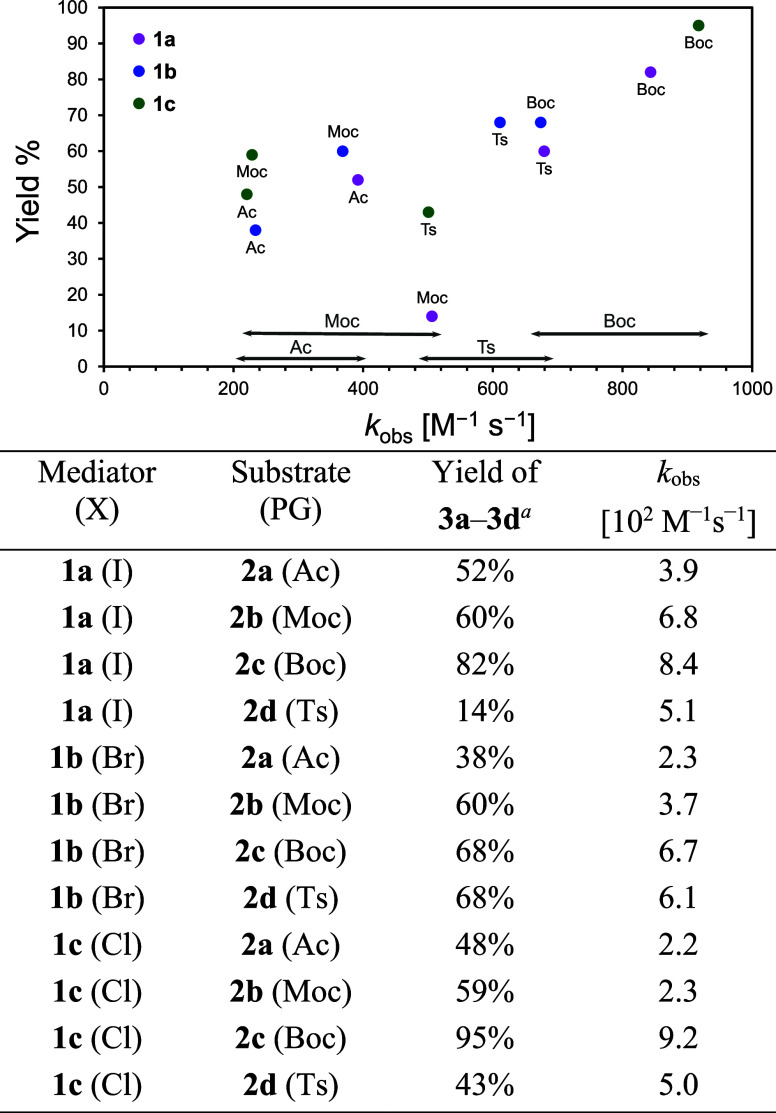 4
4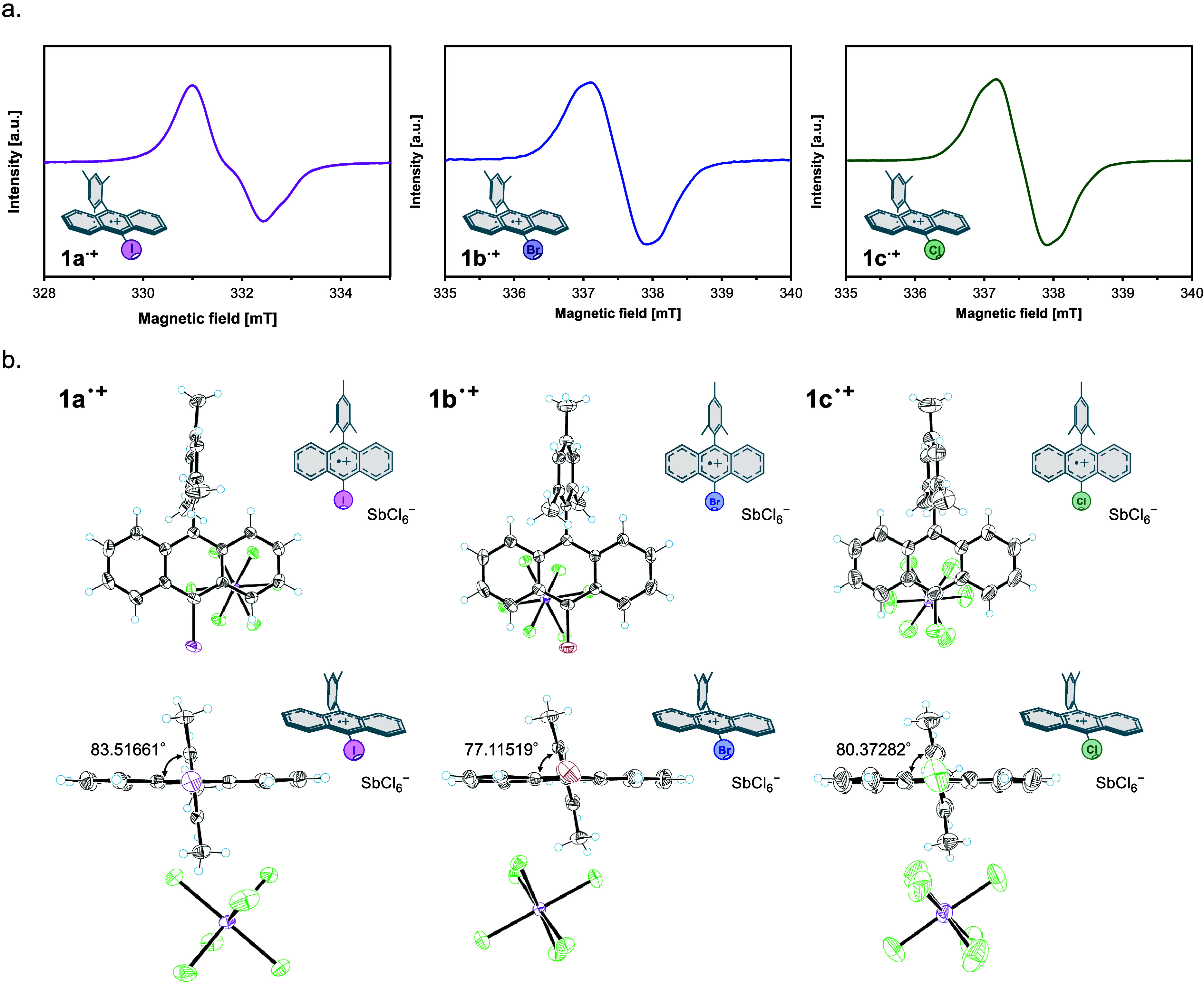 5
5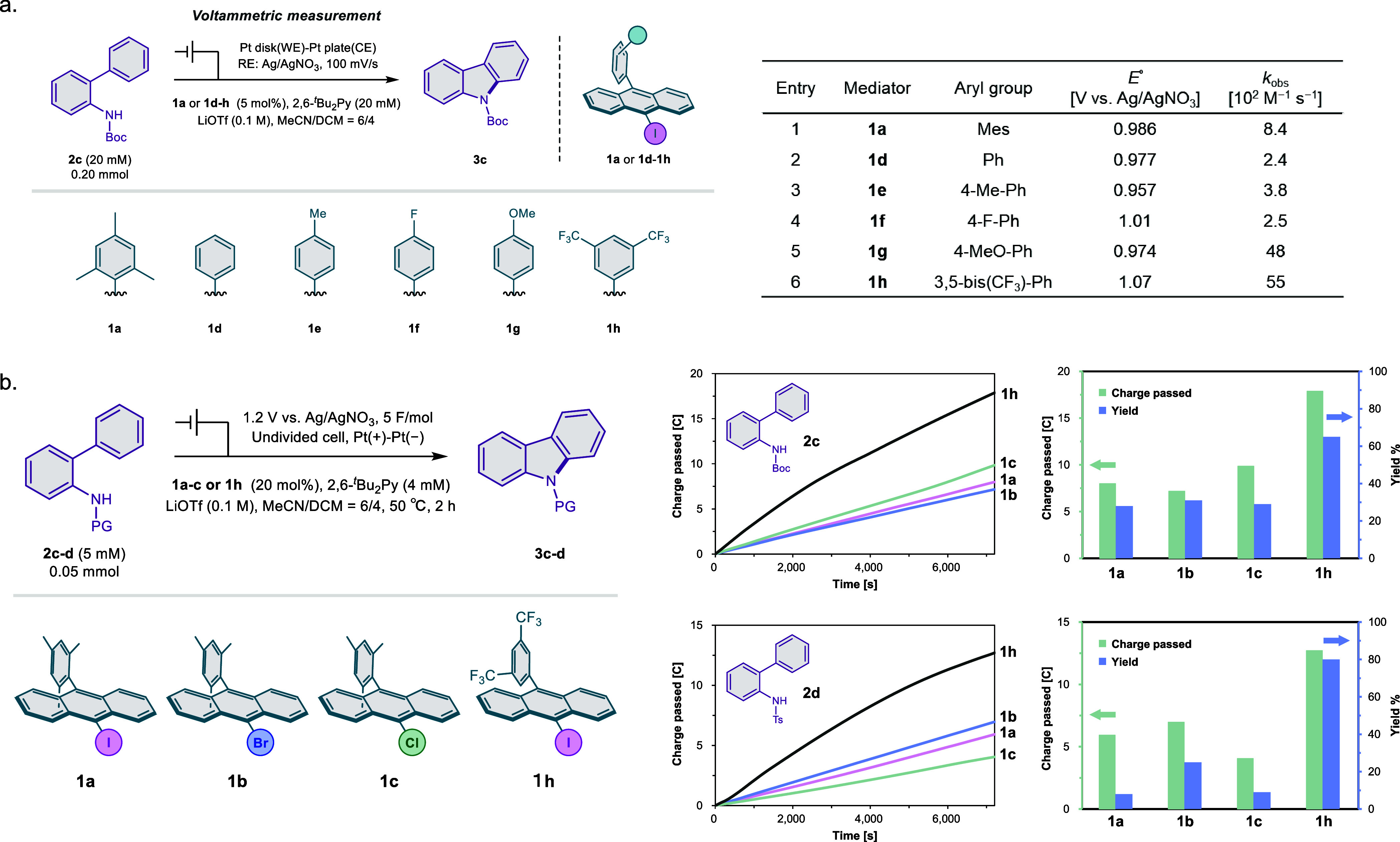 6
6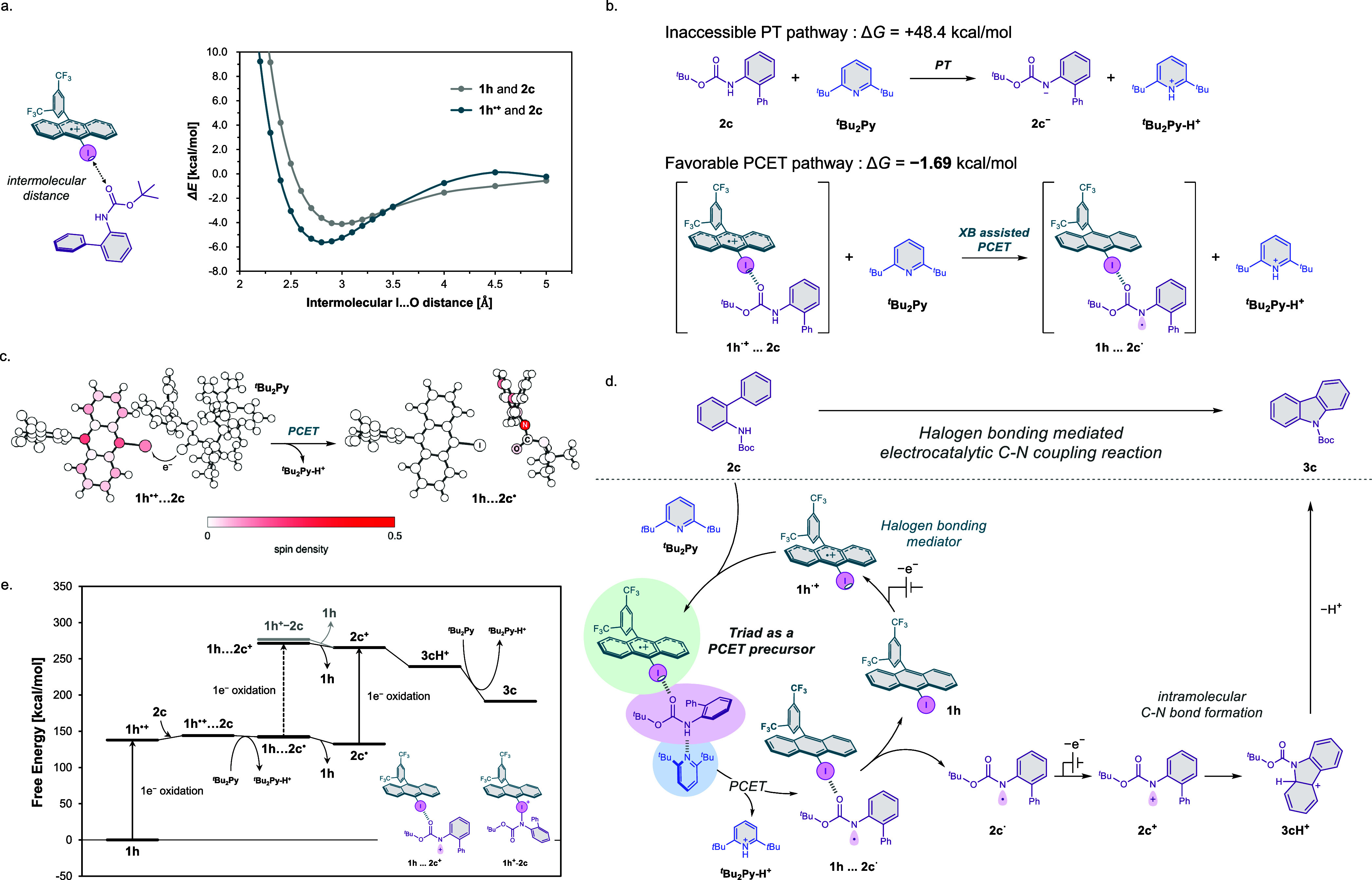 7
7- —JSPS KAKENHINA
- —JSPS KAKENHINA
- —JSPS KAKENHINA
- —JSPS KAKENHINA
- —JSPS KAKENHINA
- —Murata Science FoundationNA
- —Sasakawa Scientific Research Grant, Japan Science SocietyNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRadical Photochemical Reactions · Vanadium and Halogenation Chemistry · Metal-Catalyzed Oxygenation Mechanisms
Introduction
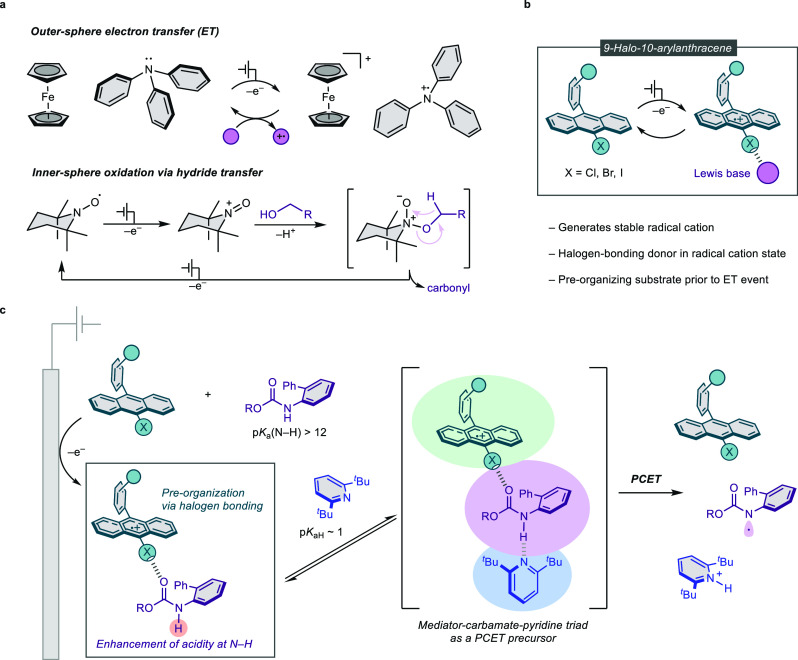
Redox mediators are essential components in electrocatalysis, facilitating electron transfer between electrodes and substrates while often enabling reactions under milder and more selective conditions. ?,? Their role becomes particularly critical in organic electrosynthesis, where direct electrolysis can lead to undesired side reactions due to high overpotentials, electrode fouling, or limited control over molecular recognition. ?−? ? ? ? Over the past decades, a wide variety of redox mediators, including ferrocene, triarylamines, and nitroxyl radicals, have been developed to overcome these limitations (Figurea). ?,?−? ? However, these systems predominantly rely on either outer-sphere electron transfer or the formation of covalent intermediates, which often lack dynamic, substrate-specific interaction capabilities.
Conceptual overview of this work. (a) Comparison of redox mediators by mode of substrate interaction and electron transfer. (b) Design of redox-switchable haloanthracene mediators enabling halogen bonding upon oxidation. (c) Mechanistic proposal involving halogen-bonding-induced preorganization and PCET.
In contrast, noncovalent interactions, such as hydrogen bonding and π–π stacking, offer a means of transient substrate association, but their integration into redox mediator design remains underexplored. Among them, halogen bondinga directional, tunable interaction involving polarized halogen atomshas recently emerged as a powerful tool in molecular recognition and catalysis. ?−? ? ? ? Yet, its application in the context of redox catalysis, especially as a redox-switchable interaction, has not been realized. Although several redox-active scaffolds, such as ferrocene and tetrathiafulvalene derivatives, are known to exhibit switchable halogen-bonding behavior upon electrochemical oxidation, these systems have not been utilized as mediators to control electron or proton transfer in catalytic transformations. ?−? ? ?
Herein, we report a new class of redox mediators based on haloanthracene derivatives, which exhibit stronger halogen bonding upon one-electron oxidation to stable radical cation states (Figureb). This redox-triggered halogen bonding allows for dynamic substrate capture and spatial preorganization, which, in turn, promotes efficient intramolecular C–N bond formation via electrocatalysis (Figurec). Quantitative kinetic evaluation using foot-of-the-wave analysis (FOWA), ?,? spectroscopic and structural characterization of the radical cations, and theoretical studies provide a comprehensive mechanistic understanding of this system. ?,? This work presents a new strategy in redox mediator design where redox-switchable noncovalent interactions are leveraged to control molecular proximity, electron transfer, and reaction pathways.
Results and Discussion
Design and Synthesis of Haloanthracene Mediators
We aimed to develop a mediator capable of electrochemically switchable halogen bonding, enabling a controllable substrate association under anodic conditions. Such a mediator requires a molecular framework that becomes sufficiently electron-deficient upon one-electron oxidation, thereby increasing the electrophilicity of the adjacent halogen atom. Stabilization of the oxidized electron-deficient state is crucial, as the enhanced σ-hole on the halogen must persist long enough to mediate substrate activation while avoiding undesirable side reactions. To meet these requirements, we focused on scaffolds capable of delocalizing the unpaired electron and the cation charge through an extended π-conjugation.
Less-conjugated aromatic systems such as benzene and naphthalene were expected to form unstable radical cations, whereas excessive π-extension was anticipated to lower the oxidation potential, making subsequent one-electron transfer from the substrate difficult. Based on these considerations, anthracene, phenanthrene, and pyrene, which possess comparable degrees of ring fusion, were evaluated as candidate frameworks. Cyclic voltammetry (CV) studies revealed that the anthracene framework provides the most favorable balance between radical cation stability and oxidation potential, establishing it as the optimal platform for constructing haloanthracene-based redox mediators (Figure S4).
Guided by this design, we synthesized a series of haloanthracene-based mediators bearing different halogen substituents (I, Br, Cl) at the 9-position (Figurea). The anthracene core was functionalized with a bulky mesityl group at the 10-position to enhance solubility and sterically protect the radical cation state. 9-Halo-10-mesitylanthracenes were obtained in three variantsiodo (1a), bromo (1b), and chloro (1c)via electrophilic halogenation of a common 10-mesitylanthracene precursor under mild conditions (see Supporting Information for details). Their structures were confirmed by NMR and HRMS analyses.
(a) The designs of haloanthracene-based redox mediators and their redox potentials and peak separations. (b) Redox behavior of 1a–1c. Cyclic voltammograms of 1 mM 1a–1c in 0.1 M LiOTf/MeCN + CH2Cl2 (6:4 in vol) at a scan rate of 0.1 V s–1.
CV measurements showed quasi-reversible oxidation waves for all three mediators, corresponding to the formation of their radical cation states (1a ^ •+ ^–1c ^ •+ ^) (Figureb). The formal oxidation potentials (E°) showed only modest variation: 0.99 V vs Ag/AgNO_3_ for 1a and slightly higher values around 1.05 V for 1b and 1c. These results indicate that the halogen substituent exerts only a minor influence on redox potential, while all three mediators exhibit sufficient electrochemical reversibility (ΔE p = 95–99 mV) and stability to operate under the electrocatalytic conditions explored in this study.?
Application to Intramolecular C–N Bond Formation
To evaluate the catalytic performance of the haloanthracene mediators, we applied them to the intramolecular oxidative cyclization of a series of N-protected 2-aminobiphenyl derivatives. The reaction conditions were systematically optimized, and the full set of screening data and discussion of their implications are provided in the Supporting Information (Section 3–5). Four substrates with different nitrogen protecting groups were examined: acetyl (Ac: 2a), methoxycarbonyl (Moc: 2b), tert-butoxycarbonyl (Boc: 2c), and tosyl (Ts: 2d). These substrates undergo two-electron oxidation to furnish the corresponding carbazole products (3a–3d). ?,? The reaction proceeded successfully with several aminobiphenyl derivatives bearing diverse substituents, suggesting that the catalytic system can accommodate various functional groups (see Supporting Information, Section 2–19).
Electrolysis was conducted in an undivided cell equipped with platinum electrodes and a Ag/AgNO_3_ reference electrode. Reactions were performed under a constant potential of 1.2 V vs Ag/AgNO_3_ in MeCN/CH_2_Cl_2_ with 0.1 M lithium triflate (LiOTf) as the supporting electrolyte. Each experiment contained 5 mM substrate, 1 mM mediator, and 4 mM 2,6-di-tert-butylpyridine (^ t ^Bu_2_Py) as the base. Electrolysis was performed at 50 °C until 5 F mol^–1^ of charge was passed.
The catalytic performance of iodoanthracene mediator 1a was first evaluated across the four substrates (Figure). Efficient C–N bond formation was observed in all cases, with yields of 52% for 2a (Ac), 60% for 2b (Moc), 82% for 2c (Boc), and 14% for 2d (Ts). The highest efficiency was seen with the Boc-protected substrate, while the Ts group significantly suppressed the reactivity. The lower reactivity is attributed to its strong electron-withdrawing nature and poor charge delocalization, resulting in insufficient intermediate stabilization.?
Intramolecular C–N bond formation reaction of various N-protected aminobiphenyls.
For comparison, bromo (1b) and chloro (1c) mediators were also tested. In most cases, 1a delivered product yields that were comparable to or higher than those obtained with 1b and 1c. For example, with 2a (Ac) and 2b (Moc), 1a afforded the highest yields (52% and 60%, respectively). In the case of 2c (Boc), although 1c gave the highest yield (95%), 1a also performed well (82%). For 2d (Ts), 1a gave a lower yield (14%) compared to 1b and 1c. These results suggest that 1a shows a particularly favorable performance with moderately electron-withdrawing protecting groups such as Boc and Moc. This trend hints at potential noncovalent interactions, such as redox-induced halogen bonding, that may enhance reactivity under specific substrate electronic environments.
Quantitative Kinetic Evaluation via Foot-of-the-Wave Analysis
(FOWA)
To gain deeper insights into the reactivity trends observed in electrocatalytic C–N bond formation, we conducted FOWA to determine apparent rate constants (k obs) for each mediator–substrate combination. FOWA is a widely used electrochemical method that enables kinetic analysis of catalytic systems by extracting rate constants from the rising portion (“foot”) of a catalytic wave in cyclic voltammograms. ?−? ? ? ? It offers the advantage of requiring only minimal catalytic current, allowing reliable comparison under steady-state conditions even in the presence of diffusion limitations.?
The results are summarized in Figure. 1a (X = I) consistently exhibited higher k obs values across almost all of the substrates, indicating its superior ability to facilitate electron transfer under catalytic conditions. For instance, with 2c (Boc), 1a achieved the highest rate constant of 8.4 × 10^2^ M^–1^ s^–1^, followed by 6.8 × 10^2^ M^–1^ s^–1^ with 2b (Moc) and 3.9 × 10^2^ M^–1^ s^–1^ with 2a (Ac). Even for the more challenging Ts-substituted substrate 2d, 1a maintained a moderate k obs of 5.1 × 10^2^ M^–1^ s^–1^. In contrast, bromo-substituted mediator 1b displayed higher overall rate constants. The chloro variant 1c showed a similar or slightly lower trend. These observations underscore the kinetic advantage of the iodo-substituted system, which is attributed to its high polarizability and redox-stable radical cation state that likely enhances substrate preorganization via halogen bonding.? Among the protecting groups examined, the Boc group (2c) consistently exhibited the most favorable reactivity. Moreover, a clear positive correlation between the rate constant (k obs) and the product yield was observed across all substrates (2a–2d), indicating that the kinetic efficiency of the mediators directly reflects their effectiveness in promoting product formation.
Yield vs k obs plot (top) and table (bottom) of intramolecular C–N bond formation reaction using haloanthracene mediators 1a–1c.
Spectroscopic and Structural Characterization of Radical Cation
Intermediates and Their Interaction with a Lewis Base
CV studies of 1a–1c revealed reversible oxidation waves, indicating that radical cation species (1a ^ •+ ^–1c ^ •+ ^) can be stable enough to be characterized. To confirm the generation of these intermediates under catalytic conditions, EPR spectroscopy was performed in the same electrolyte solution used for electrocatalysis (Figurea). In all cases, characteristic EPR signals attributable to radical species were observed, consistent with the formation of persistent π-radical cations.
*(a) EPR spectrum of 1a
•+ (top left) and 1b
•+ (top middle) and 1c
•+ (top right). (b) X-ray structures of the SbCl6 – salts of 1a
•+ and 1b
•+ at 150 K and 1c
•+ at 200 K. The thermal ellipsoids are shown in 50% probability and the solvent molecules (CH2Cl2 and Et2O) are omitted for clarity.*
We further succeeded in the chemical oxidation of 1a–1c using tris(2,4-dibromophenyl)ammoniumyl hexachloroantimonate (magic green) as a one-electron oxidant. The resulting radical cation salts were purified and subjected to slow crystallization at −20 °C, which allowed us to isolate single crystals of all three radical cations (1a ^•+^–1c ^•+^). To the best of our knowledge, this represents the first crystallographic characterization of haloanthracene radical cations.
The solid-state structures revealed that the anthracene π-framework remains largely planar upon oxidation with only minor changes in bond lengths and angles across the three systems. This structural rigidity indicates that the π-radical is well delocalized and structurally stable, supporting its suitability as a one-electron redox mediator. The mesityl substituent was found to adopt a nearly orthogonal orientation relative to the anthracene core, sterically shielding the π-surface and likely contributing to the stability of the radical cations.
Qualitative observations during crystallization and handling revealed that differences in chemical stability were as follows. 1a (I) exhibited the highest persistence under ambient conditions, while 1c (Cl) degraded most rapidly, suggesting that the heavier halogen imparts greater oxidative stability.
We have also performed voltammetric analysis to clarify the presence of halogen-bonding interactions between 1a and Lewis base (see Supporting Information, Section 3.4 for detail). Square wave voltammetry of 1a ^ •+ ^ was recorded with successive addition of a halogen-bond acceptor, ClO_4_ ^–^ anion, leading to systematic shifts in the oxidation potential. This result indicates that a more Lewis basic substrate such as 2a can readily interact with 1a ^ •+ ^. In addition, the yield of 3a was significantly decreased when nonhalogen-bonding analogues, 9-methyl-10-mesitylanthracene (1i) and 9-methyl-10-(3,5-bis(trifluoromethyl)phenyl)anthracene (1j), were used as mediators (see Supporting Information, Section 3.6).
Structural Diversification of Iodoanthracene Mediators
Encouraged by the promising results of bulk electrolysis, kinetic analysis, and structural characterization, we further explored the structural tunability of the iodoanthracene scaffold. To this end, we synthesized a series of derivatives in which the 10-position mesityl group of the parent mediator 1a was replaced with other aryl groups to modulate the electronic and steric environment (Figurea). Specifically, we prepared five new derivatives, 1d–1h. These compounds maintain the core 9-iodoanthracene structure and allow systematic evaluation of aryl substituent effects on mediator performance.
(a) Voltammetric kinetic analysis of 9-Iodo-10-arylanthracene (1a and 1d–1h) (top left). E° of 1a and 1d–1h and yield and k obs of the reaction are shown in the top-right table. (b) Q–t plots in the C–N bond formation of 2c–2d using 1a–1c and 1h and charge passed and yield in the C–N bond formation (bottom right).
Using 2c as a model substrate, we performed FOWA to determine the apparent rate constants (k obs) for each new mediator (Figurea). The results revealed large variations in catalytic activity depending on the aryl substituent. These results indicate that 1h exhibits exceptional catalytic activity, surpassing that of the original 1a by nearly an order of magnitude. This enhancement is likely due to the highly electron-withdrawing nature of the bis(trifluoromethyl) groups, which increase the halogen-bond-donor ability.
To confirm this kinetic advantage under preparative conditions, we conducted bulk electrolysis using 1a–1c and 1h with substrates 2c (Boc) and 2d (Ts) (Figureb). Under constant potential electrolysis (1.2 V vs Ag/AgNO_3_), the total charge passed was monitored over time. The 1h-mediated reactions exhibited the fastest current response, indicating significantly accelerated catalytic turnover. Upon completion of electrolysis, the target carbazole products were obtained in high yields 65% 3c (from 2c, 3.7 F mol^–1^) and 80% 3d (from 2d, 2.6 F mol^–1^). These results confirm that aryl substitution at the 10-position of the iodoanthracene core can dramatically influence mediator activity.
Computational Analysis of Halogen Bonding and PCET Pathway
To elucidate the origin of the high reactivity exhibited by the iodoanthracene-based mediators, we conducted detailed computational studies focusing on halogen-bonding interactions and the proton-coupled electron transfer (PCET) mechanism. These studies employed density functional theory geometry optimizations at the (U)CAM-B3LYP/def2-SVP level, followed by high-level single-point energy calculations using DLPNO-CCSD(T)/ma-def2-SVP (Figurea,b,d), allowing us to construct a reliable energy profile for the full two-electron oxidation process. To evaluate the solvation effect, the RISM-SCF-cSED approach, which is one of the hybrid methods between quantum mechanics and statistical mechanics, was employed. In the energy diagram, the thermal correction was included by employing vibrational frequency analysis. The computational details are summarized in Supporting Information.
*(a) Potential energy curves for 1h and 1h
•+ complexed with 2c. (b) ΔG comparison between the PT process and halogen bonding (XB)-assisted PCET process. (c) Spin densities of 1h···2c
• interacted with t Bu2Py-H+ and (top) and 1h···2c
• (bottom). (d) Plausible reaction mechanism. (e) Energy diagram of the electrocatalytic C–N coupling reaction.*
Our first objective was to assess the presence of halogen-bonding interactions between the mediators and the substrate. Using 2c as a model substrate, we systematically varied the distance between the halogen atom of the mediator 1h and the carbonyl oxygen of the substrate and evaluated the interaction energies in both neutral and oxidized states (Figurea). In their neutral forms, the mediators exhibited only weak interactions with the substrate. However, upon one-electron oxidation, the interaction was strengthened, as evidenced by the appearance of a relatively lower local minimum in the energy curves at X···O distances shorter than the sum of the van der Waals radii (3.20–3.55 Å).? These results indicate that halogen bonding becomes significantly more favorable in the radical cation state. These interactions were observed consistently across all four mediators studied (Figure S32).
Next, we examined the thermodynamics of the abstraction of proton from the substrate by ^ t ^Bu_2_Py. Given the approximate pK a of more than 12 for the N–H moiety of N-acyl aniline, which possesses electronic properties comparable to those of the substrate, and the pK aH of around 1 for the base, such deprotonation is thermodynamically uphill in the absence of mediation. ?,? This expectation was confirmed by calculations of the deprotonation free energy of 2c with and without the radical cation of 1h. The deprotonation free energies are +48.4 kcal/mol without 1h ^ •+ ^ and −1.7 kcal/mol with 1h ^ •+ ^. By taking the complex form between 2c and 1h ^ •+ ^, deprotonation by the weak base from the N–H moiety of N-acyl aniline becomes possible.
The large stabilization in the deprotonation process with 1h ^ •+ ^ is caused by the PCET. Performing the spin density analyses further clarified the PCET nature of the process (Figurec). When the resulting pyridinium ion remained in the vicinity of the mediator-substrate complex, the unpaired electron was localized on the anthracene moiety of the mediator. In contrast, when the pyridinium ion was computationally removed, the spin density shifted onto the substrate. This redistribution implies that the hydrogen bond plays a critical role in stabilizing the charge and spin on the mediator during the PCET event and that the departure of the base triggers electron transfer to the substrate. These findings are fully consistent with a PCET mechanism. ?−? ?
To contextualize these mechanistic insights, we proposed a plausible mechanism (Figured) and constructed an energy diagram for the two-electron oxidation process (Figuree). The initial one-electron oxidation leads to the generation of a mediator radical cation that forms a halogen-bonded complex with the substrate in its oxidized form. The base also interacts with this complex to yield the key intermediate in the PCET step. After the PCET, the resulting halogen-bonded complex 1h···2c ^ • ^ can either dissociate to give the free radical 2c ^ • ^ or undergo direct oxidation while remaining associated. The oxidation energies of both 1h···2c ^ • ^ and free 2c ^ • ^ are both smaller than that of the first oxidation step (1h → 1h ^ •+ ^), indicating that the second oxidation is energetically feasible through either pathway. On the other hand, the dissociation of 1h···2c ^ • ^ is exothermic, making it more likely that the second oxidation occurs after the dissociation event. Cationic species 2c ^ + ^ generated by the second oxidation can interact with the mediator 1h to form a halogen-bonding complex 1h···2c ^ + ^ and a hypervalent state 1h ^ + ^ –2c. Such hypervalent iodine species are well-documented to engage in C–N or C–O coupling reactions, suggesting that analogous reactivity could be operative in this system. ?,?,?−? ? However, because both complexes are not stable compared to the free form, the complex form is not important after the 2c ^ + ^ is produced. Intramolecular ring formation of 2c ^ + ^ takes place to afford 3cH ^ + ^ and 3c is obtained after deprotonation by ^ t ^Bu_2_Py.
Taken together, these computational findings provide strong support for the proposed mechanism involving redox-switchable halogen bonding and PCET. This mechanistic framework underpins the observed high reactivity and selectivity, particularly for optimized mediator 1h, and provides a clear design rationale for future development of advanced redox mediators.
Conclusions
In summary, we have developed a new class of redox mediators based on haloanthracene derivatives that exhibit redox-switchable halogen bonding in their radical cation states. These mediators enable efficient electrocatalytic intramolecular C–N bond formation through enhanced substrate preorganization and activation via noncovalent interactions. Among them, iodoanthracene mediator 1a demonstrated broad substrate compatibility and favorable kinetics, which were further improved through structural tuning at the 10-position.
Systematic modification of the aryl substituent at the 10-position of the iodoanthracene scaffold revealed that the 3,5-bis(trifluoromethyl)phenyl-substituted derivative 1h significantly outperformed its analogs, as evidenced by kinetic analysis and bulk electrolysis experiments. The pronounced enhancement in reactivity was traced to its ability to form stronger halogen-bonding interactions in the oxidized state.
Computational studies provided detailed mechanistic insights, confirming the emergence of halogen bonding only upon one-electron oxidation and demonstrating that this interaction promotes a PCET pathway by increasing the acidity of the substrate’s N–H bond.
This work highlights a new design principle for redox mediators that leverages redox-responsive noncovalent interactions to control both molecular recognition and electron–proton dynamics. The concept of halogen-bonding-assisted PCET opens new directions in electrocatalyst design, offering a broadly applicable strategy for enhancing selectivity and efficiency in synthetic electrochemistry.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Francke R.Little R. D.Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments Chem. Soc. Rev.20144382492252110.1039/c 3cs 60464 k 24500279 · doi ↗ · pubmed ↗
- 2Shao W.Lu B.Cao J.Zhang J.Cao H.Zhang F.Zhang C.The Use of Redox Mediators in Electrocatalysis and Electrosynthesis Chem.Asian J.2023182 e 20220109310.1002/asia.20220109336577711 · doi ↗ · pubmed ↗
- 3Wang F.Stahl S. S.Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron-Proton Transfer Mediators Acc. Chem. Res.202053356157410.1021/acs.accounts.9b 0054432049487 PMC 7295176 · doi ↗ · pubmed ↗
- 4Hossain M. M.Aldous L.Polyoxometalates as Solution-Phase Electrocatalytic Mediators for Reduced Electrode Fouling and the Improved Oxidative Response of Phenols Electrochem. Commun.201669323510.1016/j.elecom.2016.05.020 · doi ↗
- 5Medici F.Resta S.Andolina S.Benaglia M.Recent Advances in Enantioselective Catalytic Electrochemical Organic Transformations Catalysts 202313694410.3390/catal 13060944 · doi ↗
- 6Novaes L. F. T.Liu J.Shen Y.Lu L.Meinhardt J. M.Lin S.Electrocatalysis as an Enabling Technology for Organic Synthesis Chem. Soc. Rev.202150147941800210.1039/D 1CS 00223 F 34060564 PMC 8294342 · doi ↗ · pubmed ↗
- 7Chen N.Wu Z.-J.Xu H.-C.Ferrocene as a Redox Catalyst for Organic Electrosynthesis Isr. J. Chem.2024641–2e 20230009710.1002/ijch.202300097 · doi ↗
- 8Nutting J. E.Rafiee M.Stahl S. S.Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions Chem. Rev.201811894834488510.1021/acs.chemrev.7b 0076329707945 PMC 6284524 · doi ↗ · pubmed ↗