Exploring the Synergistic Effects of Inactive Materials and Processing on Aqueous Fabrication of Poly(3‐Vinyl‐N‐Methylphenothiazine) Positive Electrodes for Lithium‐Organic Batteries
Sathiya Priya Panjalingam, Philipp Penert, Markus Börner, Birgit Esser, Martin Winter, Peter Bieker

TL;DR
This study explores eco-friendly aqueous methods to fabricate electrodes for organic batteries, using water-based binders and optimizing processing parameters to achieve high performance.
Contribution
The paper introduces a novel aqueous processing method for poly(3-vinyl-N-methylphenothiazine) electrodes using water-processable binders and identifies key factors affecting performance.
Findings
Electrodes using Na-CMC retained ≈90% of theoretical capacity over 1000 cycles at 1C rate.
SBR co-binder improved mechanical integrity and mitigated negative effects of high densification pressure.
Processing factors like conductive additive and mixing method strongly influence electrochemical performance.
Abstract
Organic redox‐active electrode materials are gaining increasing attention due to their eco‐friendliness, abundance, and structural versatility. However, their processing typically depends on poly(vinylidene difluoride) (PVdF) as binder and N‐methyl‐2‐pyrrolidone (NMP) as solvent, both are expensive and hazardous. While aqueous processing methods are well established for inorganic electrodes, their application to organic materials remains largely unexplored. This study investigates the use of water‐processable binders, specifically sodium carboxymethyl cellulose (Na‐CMC) and styrene‐butadiene rubber (SBR) for fabricating poly(3‐vinyl‐N‐methylphenothiazine) electrodes. Key factors influencing electrode performance and microstructure were systematically studied, including the choice of conductive additive, mixing procedures, hot‐pressing, and densification. Among these, the selection of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 FIGURE 1
FIGURE 1 FIGURE 2
FIGURE 2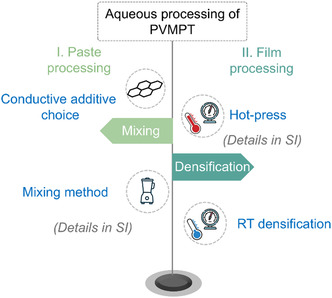 FIGURE 3
FIGURE 3 FIGURE 4
FIGURE 4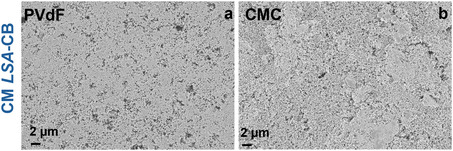 FIGURE 5
FIGURE 5 FIGURE 6
FIGURE 6 FIGURE 7
FIGURE 7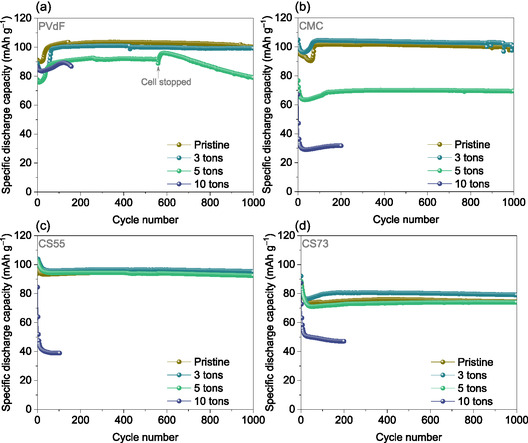 FIGURE 8
FIGURE 8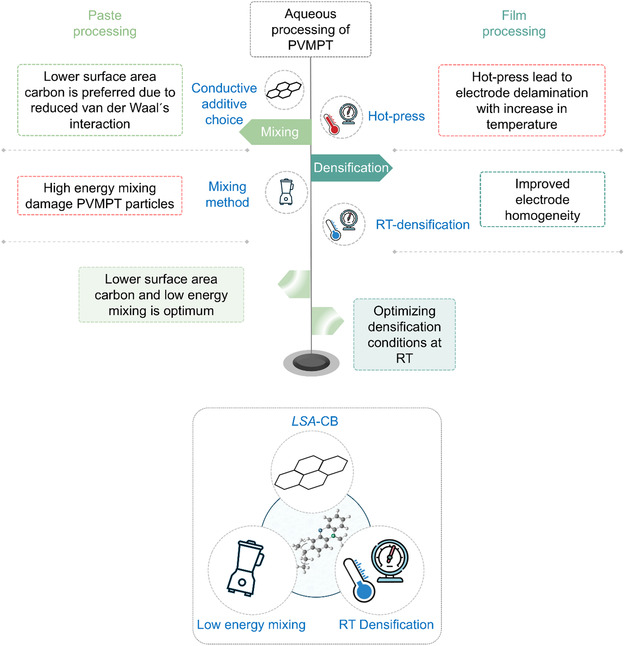 FIGURE 9
FIGURE 9| Binders, wt% | CMC | CS55 | CS73 |
|---|---|---|---|
| CMC | 100 | 50 | 70 |
| SBR | — | 50 | 30 |
| Notations of the mixing procedure | Mixing procedure | Operating conditions |
|---|---|---|
| HEM 1 | High‐frequency mixing | 30 Hz−2 h |
| HEM 2 | Initial deagglomeration with binder and CB, followed by PVMPT addition with low‐frequency mixing |
30 Hz−1 h; 15 Hz−15 min |
| HEM 3 |
Deagglomeration with binder and CB, followed by PVMPT addition with high‐frequency mixing |
30 Hz−1 h; 20 Hz−20 min; 30 Hz−10 min |
|
Mixing method | Electrode components | Densification parameters | Notations for densification |
|---|---|---|---|
| CM method | CM |
Pristine, 3, 5 and 10 tons | PVdF |
| CM | CMC | ||
| CM | CS55 | ||
| CM | CS73 |
- —Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein‐Westfalen
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvancements in Battery Materials · Conducting polymers and applications · Advanced Battery Materials and Technologies
Introduction
1
Recent advancements in electrode manufacturing have introduced biopolymers as cost‐effective and sustainable alternative binders for lithium ion batteries (LIBs), with water‐processable cellulose derivatives emerging as strong competitors to fluorinated binders. Traditionally, poly(vinylidene difluoride) (PVdF) is the commonly used binder, while N‐methyl‐2‐pyrrolidone (NMP) serves as the processing solvent and dispersant for positive electrode production. However, PVdF is a hazardous mutagenic compound, and NMP is a toxic, flammable, and teratogenic processing solvent, both contribute to high production costs and environmental concerns [1, 2, 3, 4]. To address these challenges, sodium carboxymethyl cellulose (Na‐CMC), a bio‐derived, water‐processable binder, has been widely adopted, often in combination with styrene‐butadiene rubber (SBR), for the fabrication of LIB negative electrodes [1, 2, 4]. CMC serves as both a thickener and a dispersant/surfactant, while SBR enhances binding properties, mechanical stability, flexibility, and heat resistance [1, 3, 5]. Although binders constitute only a small fraction (2–5 wt%) of the total electrode composition, they play a crucial role in overall battery performance [1, 3, 4]. While they do not directly contribute to electrochemical activity, binders establish a well‐connected network between the active material and conductive additives (cohesion) while ensuring strong adhesion of the composite electrode to the current collector [1, 2, 3, 4]. This structural integrity is essential in preventing electrode disintegration under the mechanical and chemical stresses of continuous charge–discharge cycling [3]. Selecting the appropriate binder and optimizing its weight ratio is crucial to addressing these challenges. An ideal binder should not only facilitate uniform dispersion but also be cost‐effective, environmentally friendly, and exhibit high mechanical flexibility, thermal stability, and electrochemical resilience over the required potential range [2, 4]. While aqueous electrode processing offers several advantages, including eco‐friendliness and cost‐effectiveness, it also presents multiple challenges. These include the agglomeration of conductive additives and active materials in water due to strong electrostatic interactions and hydrogen bonding, corrosion of aluminum current collectors, insufficient wetting caused by water's high surface tension, leaching of lithium ions and transition metals, and the presence of residual moisture in the electrodes, requiring additional drying steps [2, 4]. Despite these challenges, overcoming these obstacles is critical for the successful adoption of water‐based processing in advanced electrode fabrication. Additionally, the physicochemical interactions of the solid components in aqueous electrode pastes are often overlooked, yet they substantially influence electrochemical performance. For instance, carbon black particles tend to form agglomerates, disrupting the well‐connected network and ultimately reducing cycle life and rate capability [6].
Redox‐active organic materials have attracted attention as promising electrode materials for next‐generation energy storage due to their potential sustainability, low cost, abundance, and relatively high specific capacities [7, 8, 9, 10]. Their structural flexibility enables both n‐type and p‐type charge storage, allowing for diverse cell configurations [8, 11, 12]. Among these materials, redox polymers stand out due to their excellent cycling stability, high‐rate capability, negligible solubility in electrolytes, and high specific capacities [7, 13, 14]. Despite these advantages, most reported organic electrodes still rely on PVdF binders [15, 16]. While some studies have explored aqueous binders for electrode processing [17, 18, 19], only a few specifically focus on the aqueous processing of water‐soluble organic electrode materials [20, 21]. However, redox polymers, due to their inherent insolubility in water, present considerable challenges for water‐based processing, an area that remains largely underexplored.
This study investigates the challenges and potential solutions for aqueous processing of organic redox polymer electrodes. Specifically, it examines the electrochemical performance of the redox polymer poly(3‐vinyl‐N‐methylphenothiazine) (PVMPT) when formulated with eco‐friendly, water‐processable binders, CMC and SBR, at varying weight percentages. PVMPT is a p‐type, nonconjugated heterocyclic redox polymer featuring a phenothiazine redox moiety grafted as a side chain onto an aliphatic polymer backbone. It is known for its outstanding cycle life, high‐rate capability, reasonable theoretical specific capacity (112 mAh g^−1^) considering a one‐electron redox, and an average discharge potential of 3.5 V vs. Li|Li^+^ [15, 16, 22, 23, 24]. Special attention is given to optimizing each processing step, including the selection of conductive additive, mixing techniques to improve homogeneity, and densification conditions to improve electrode homogeneity and electrochemical performance.
Experimental Section
2
Different Composition of Binders Used in This Study
2.1
This study explores two primary binder systems (PVdF and CMC) and three binary binder systems, composed of CMC and SBR and formulated in three different proportions, as detailed in Table 1. The PVdF electrode system serves as a reference to compare with other aqueous‐based systems. To ensure uniform mixing with water, all aqueous binders were prepared as solutions.
Synthesis of PVMPT
2.2
PVMPT was synthesized as previously reported [22]. Powder X‐ray diffraction (XRD), Fourier transform‐infrared spectroscopy (FT‐IR), Raman spectroscopy, and Scanning electron microscopy (SEM) were used to characterize the as‐synthesized polymer including phase identification, nature of bonds, and the morphology of the polymer.
Electrode Preparation
2.3
Electrodes were fabricated by blending the synthesized PVMPT powder with commercially available conductive additives—low surface area carbon black (LSA‐CB) (Super C65, Imerys Graphite & Carbon) and high surface area carbon black (HSA‐CB) (Ketjenblack EC‐300J, Fuel cell store), along with PVdF (Solef 5130, Solvay), sodium carboxymethyl cellulose (Na‐CMC, CRT 2000 PPA12, Dow Wolff Cellulosics, Germany), and SBR (Synthomer, 50% SBR, 50% H_2_O). These components were combined in a weight ratio of 50:45:5. Electrode paste preparation was carried out using two different mixers: a Thinky mixer and a Swing Mill mixer, with the choice of conductive additives for binder optimization. LSA‐CB refers to low surface area carbon black, while HSA‐CB refers denotes high surface area carbon black.
Thinky Mixer Electrode Paste Preparation (CM Method)
2.3.1
For electrode paste preparation using the Thinky mixer with LSA‐CB and HSA‐CB conductive additives, nonaqueous electrode pastes (PVdF‐based) were prepared by blending PVMPT, conductive additives, and PVdF (in the ratios PVMPT:LSA‐CB:PVdF and PVMPT:HSA‐CB:PVdF) with the required amount of NMP (battery grade, Sigma Aldrich). The mixture was processed using a Thinky mixer (Arm‐310) at 1700 rpm for 1 h. However, this method was ineffective for aqueous electrode pastes, as the material did not adequately disperse in the Thinky mixer alone. To overcome this, the solid components were initially hand‐mixed using a mortar and pestle for 30 min. The binder solution and the required amount of water were then added, followed by further mixing in the Thinky mixer at 1700 rpm for 1 h until the desired electrode paste consistency was achieved. The Thinky mixer mixing is designated as CM method, because this involves centrifugal mixing of the components, and so the electrodes prepared using a Thinky mixer with different conductive additives (HSA‐CB and LSA‐CB) and binders are designated as CM HSA‐CB_Binder_ and CM LSA‐CB_Binder_. The electrodes that have not undergone any densification are referred to as pristine (or non‐densified) electrodes.
Swing Mill Mixer Electrode Paste Preparation (HEM Method)
2.3.2
For electrode preparation using the MM400 Swing Mill mixer (only CM), all electrode components were added to the stainless‐steel Swing Mill jar (5 mL volume) together with two stainless steel balls (0.078 g each). In this process, the mixer jars oscillated back and forth at a fixed frequency, facilitating mixing through the collision of balls and the electrode materials moving in both directions. This swing mill mixing approach is classified as a high energy mixing (HEM) method. In this study, three distinct procedures were employed for the electrode paste preparation using the swing mill mixer. For simplicity, abbreviated labels for each method are introduced and summarized in Table 2.
The electrodes, prepared using a Swing Mill mixer with different conductive additives (HSA‐CB and LSA‐CB) and binders, are designated as HEM HSA‐CB_Binder_ and HEM LSA‐CB_Binder_. The mixing procedures are represented by HEM 1, HEM 2, and HEM 3. For ease of simplicity, all the notations used for representing electrode composition and mixing method are summarized in Table S1.
The prepared electrode pastes were spread onto 5 wt% KOH‐etched aluminum foil (Speira, 20 µm) using the doctor‐blade (Zehntner 4‐sided film applicator) method (50 µm spacing). The electrodes underwent a two‐step drying process: primary drying in a hot air oven for 1 h, followed by secondary drying in a vacuum drying cabinet at 70°C overnight. Electrode discs (12 mm in diameter) were then prepared, densified at different pressure using a hydraulic press for 20 s, and further dried under reduced pressure (0.005 mbar) in a Büchi B‐585 glass oven at 80°C for non‐aqueous discs and 120°C for aqueous discs for 12 h. The final electrodes exhibited dry film thicknesses of 10–13 µm (excluding the current collector) and active mass loadings between 0.18 and 0.26 mg cm^−2^.
Cell Assembly and Electrochemical Measurements
2.4
The electrochemical performance of the PVMPT electrodes was evaluated using a three electrode Swagelok T‐cell setup with lithium metal serving as both the counter (12 mm disc) and reference electrode (5 mm disc), while PVMPT composite electrodes as working electrode. The electrolyte used for the measurements was 1.0 m LiPF_6_ in 3:7 EC: EMC (v/v) (Solvionics, 99.9% H_2_O: 20 ppm max). Six layers of Freudenberg 2190 non‐woven polypropylene separator (FS‐2190) was soaked with 130 µL electrolyte for the counter and the working electrode (13 mm separator) while 60 µL (10 mm separator) at the reference electrode. The separators were punched and pre‐dried under reduced pressure (0.005 mbar) at 120°C for 24 h in Büchi B‐585 glass oven before cell assembly. The molecular sieves activated at 300°C for 24 h under reduced pressure were used to dry the electrolyte prior to using for the cell assembly. The cells were assembled in a dry room or glove box with humidity levels below 20 ppm, allowed to rest for 12 h before cycling. Constant current cycling (CCC) investigations were performed using a MACCOR 4000 series battery tester at 1C rate within a voltage range of 3.0–4.0 V.
Physical Characterization
3
X‐Ray Diffraction
3.1
The powder XRD pattern of the as‐synthesised PVMPT polymer was obtained by using a Bruker D8 Advance with Cu kα radiation (λ‐ 1.54 Å) in Bragg–Brentano geometry. The measurements were conducted over a 2θ range of 5°–90°, with a step size of 0.02° s^−1^ and a recording time of 2 s per step. The instrument was operated at 40 kV and 20 mA, utilizing a 0.5 m divergence slit for data acquisition.
Fourier Transform‐Infrared Spectroscopy
3.2
The FT‐IR spectrum of PVMPT was measured using a Bruker Vertex 70 in the mid IR region between 400 and 4000 cm^−1^. The measurement was performed with a single‐reflection attenuated total reflectance unit featuring a diamond crystal and a KBr beam splitter. Prior to the measurement, the chamber was purged with nitrogen (N_2_) gas for 1 h, and the spectrum was recorded under a continuous N_2_ flow.
Raman Spectroscopy
3.3
The Raman spectrum of PVMPT was recorded using a confocal microscope (Horiba Scientific, LabRAM HR Evolution) equipped with an air‐cooled charge coupled detector (detector and a 600 g mm^−1^ grating. Excitation was performed with a 633 nm red laser, delivering 10.5 mW at the objective, with the power reduced to 1.05 mW using a 10% filter. A 50× objective lens (Carl Zeiss Microscopy, 9.2 mm working distance, numerical aperture 0.5) was used to focus the laser. Raman spectra were acquired through three integrations of 35 s each. Data acquisition, analysis, and spectrometer were managed using LabSpec 6.7.1.10 software (Horiba Scientific). Prior to each measurement, the system was calibrated to the crystalline silicon peak at 520.7 cm^−1^.
Scanning Electron Microscopy
3.4
SEM micrographs of the PVMPT polymer powder and PVMPT composite electrodes were obtained using a Carl Zeiss AURIGA Crossbeam Workstation. The imaging was performed at an accelerating voltage of 3 kV with an in‐lens detector, maintaining a working distance of 4 mm. Cross‐sectional samples were prepared using a cooling cross‐section polisher (IB‐19520CCP, Jeol). The milling process included an initial coarse step at 5 kV with an argon flow rate of 2.0 for 2.5 h at a width of 100 µm, followed by a fine milling at 2 kV with an argon flow rate of 9.0 for 1 h. Energy‐dispersive X‐ray spectroscopy (analysis was conducted at an accelerating voltage of 6 kV.
Results and Discussion
4
Figure 1 illustrates the chemical structure of PVMPT, PVdF, CMC, and SBR. The gray dotted box highlights the redox‐active phenothiazine moiety.
Chemical structure of PVMPT, with the phenothiazine redox moiety highlighted by a gray dotted box, along with the binders–PVdF, CMC and SBR. PVdF = poly(vinylidene difluoride); SBR = styrene‐butadiene rubber.
Structural Characterization of as‐Synthesized PVMPT Powder
4.1
Figure 2a presents the XRD pattern of the as‐synthesized PVMPT. The pattern exhibits two broad reflections at ≈24° and 47°, corresponding to the (002) and (100)/(101) planes, confirming the polymer's amorphous nature. Amorphous materials exhibit short‐range order, imparting unique characteristics such as increased surface area, faster kinetics due to shorter percolation pathways, and greater free volume. In contrast, crystalline host structures rely on factors such as structural stability, crystal orientation, phase transitions, and limited ion accommodation sites for their performance [25, 26]. Figure 2b,c depicts the polymer's morphology at two different magnifications, revealing a non‐uniform morphology with heterogeneous particle size distribution. The presence of pores in the polymer is evident that facilitates electrolyte wettability. FT‐IR and Raman spectrum of PVMPT powder is shown in Figure S1.
Physical characterization of the synthesised PVMPT powder. (a) XRD, (b,c) SEM images at 100x and 500x magnifications respectively.
Optimization of Electrode Fabrication: Influence of Inactive Materials, Mixing Strategies, and Densification on the Electrode Microstructure and Electrochemical Performance
4.2
Herein, the electrode fabrication process is divided into two main stages: paste processing and film processing, each of which influences the electrode morphology as well as electrochemical performance. The paste processing involves the mixing and coating of electrode components. The mixing stage is particularly crucial, as it determines the homogeneity of the resulting electrode paste and electrode. Its primary goal is to break down agglomerates and ensure uniform distribution of materials. Improper mixing can lead to agglomerations which negatively affect electrochemical performance. The film processing stage includes drying and densification, which further refine the electrode structure and properties. Therefore, optimizing the formulation and processing of electrode paste is crucial for improving electrochemical performance. Figure 3 outlines the parameters investigated at the paste a well as film processing stages.
Schematic illustration of the parameters investigated at the paste as well as film processing stages. The process evolves from paste processing to film processing, with steps progressing in a top‐to‐bottom order within each stage.
In the following sections, each stage involved in both paste and film processing aimed at enhancing electrochemical performance will be discussed in detail.
Paste Processing Optimization
4.2.1
The initial step focuses on paste processing, specifically the mixing phase, which comprises two key components:
- 1.Selection of conductive additive
- 2.Mixing methods
Selection of Conductive Additive
4.2.1.1
This study first examines the impact of conductive additives and mixing method in the aqueous processing of PVMPT. Specifically, two CBs with distinct surface areas are investigated*: LSA*‐CB (59 m^2^ g^−1^) and HSA‐CB (988 m^2^ g^−1^). Electrodes incorporating HSA‐CB and LSA‐CB were prepared with two different binders, PVdF and CMC, using a Thinky mixer (low energy centrifugal mixing, hereafter referred to as the CM method).
The PVMPT electrodes fabricated using HSA‐CB combined with PVdF or CMC via the CM method are referred to as ‘CM HSA‐CB_Binder_ electrodes’. The electrodes were densified at 3 tons of pressure for 20 s. Figure 4a–c presents the surface morphology of densified CM HSA‐CB_PVdF_ and CM HSA‐CB_CMC_ electrodes and their long‐term stability at 1C rate. The pristine (non‐densified) CMC electrodes show more agglomerations (Figure S2).
Surface SEM images of CM HSA‐CB densified electrodes prepared using (a) PVdF, (b) CMC binders. Panel (c) shows the corresponding long‐term cycling stability of the electrodes evaluated at 1C rate.
Effect of Binder and Carbon Type
4.2.1.2
The electrode fabricated using a PVdF binder exhibits a more uniform and homogeneous morphology compared to the CMC‐processed electrode. This uniformity is attributed to PVMPT's solubility in NMP, the solvent used for PVdF‐based electrode processing, which enables a more even electrode microstructure. In contrast, electrodes prepared with aqueous binders show noticeable agglomerations and carbon aggregates. This inhomogeneity arises from insolubility of PVMPT in water and the cluster of carbon black aggregates forming agglomerations held together by weak physical forces such as van der Waals and electrostatic interactions in aqueous media. These effects are evident in the surface morphology and negatively affect the electrochemical performance (Figure 4c).
Building on these findings, the study further investigated whether LSA‐CB can alleviate such inhomogeneity because LSA‐CB exhibits relatively reduced van der Waals interactions.
PVMPT electrodes were therefore fabricated using LSA‐CB with PVdF, CMC binders by the same CM method (CM LSA‐CB_Binder_ electrodes), and densified at 3 tons for 20 s. Figure 5a,b displays their surface morphology. PVdF‐processed electrodes exhibit similar homogeneity regardless of the CB used, while the CMC‐processed electrodes with LSA‐CB show enhanced electrode homogeneity and reduced agglomeration compared to HSA‐CB.
Surface SEM images of densified CM LSA‐CB electrodes processed with (a) PVdF and (b) CMC binders.
The pristine (non‐densified) CM LSA‐CB and CM HSA‐CB electrodes prepared with PVdF and CMC binders are presented in Figure S2. The PVdF‐based electrodes, irrespective of the surface area of the carbon black used, display uniform morphology and well‐connected conductive network (Figure S2a and S2c). In contrast, CMC‐based electrodes display a clear dependence on carbon black type: the pristine CM LSA‐CB_CMC_ electrode presents a more homogeneous morphology (Figure S2d) than the pristine CM HSA‐CB_CMC_ electrode (Figure S2b).
This direct comparison indicates that when selecting a conductive additive for aqueous processing of redox polymers, LSA‐CB is generally advantageous.
Mixing Methods
4.2.1.3
Although LSA‐CB proves more effective overall, HSA‐CB offers several advantages, such as more active sites for ion storage (thereby enhancing charge capacity), shorter ion‐diffusion paths, and improved electrolyte retention, all of which collectively contribute to an extended cycle life. Because CM method does not ensure satisfactory homogeneity with HSA‐CB for CMC‐based processing, HEM method was explored to improve dispersion and electrode uniformity (hereafter referred to as the HEM method, see experimental section for details).
HEM method deagglomerates the particles and promotes homogeneous mixing, but electrodes produced this way exhibited limited electrochemical performance. As detailed in Supporting Information Section 1 (Figures S3 and S4), this limitation is attributed to the mechanical damage of PVMPT particles caused by high energy collision of balls inside the mixer jar. In contrast, the CM method avoids such collisions, relying on high‐speed rotation (≈1700 rpm) without any milling object, which prevents this mechanical polymer degradation.
In summary, these results indicate that LSA‐CB combined with low energy (CM method) mixing provides the most homogeneous electrode morphology, which forms the foundation for the next stage of film densification.
Film Processing: Densification
4.2.2
Following the optimized paste formulation and mixing method, the study further investigates how the film densification affects the electrode integrity and long‐term electrochemical behavior.
Film processing includes electrode densification performed under various conditions, either at elevated temperatures (hot‐pressing) and at room temperature (RT) densification under different applied pressures.
Hot‐Press
4.2.2.1
Although LSA‐CB is typically favored, *HSA‐*CB offers distinct benefits as discussed earlier. To further enhance the uniformity of CM HSA‐CB‐based electrodes, hot‐pressing was evaluated. Although hot‐pressing was expected to enhance the flexibility of PVMPT and thereby improve the uniformity of CM HSA‐CB electrodes, trials at 50°C and 70°C showed no benefit over RT densified electrodes at 3 tons. As detailed in Supporting Information Section 2 (Figure S5), higher temperature or pressure even led to electrode delamination from the current collector. Consequently, higher‐temperature experiments were discontinued and not applied to LSA‐CB electrodes, and RT densification was adopted as the practical processing strategy.
Room‐Temperature Densification
4.2.2.2
The influence of pressure on morphology was already evident from the paste‐processing stage when comparing pristine (Figure S2) and 3 ton densified electrodes (Figures 4 and 5). To systematically explore this effect, CM LSA‐CB_Binder_ electrodes combined with PVdF (reference) and various aqueous binders (CMC, CS55, and CS73) were densified at 3, 5, and 10 tons. Table 3 summarizes the mixing method, electrode compositions, densification conditions, and the corresponding notations used for RT densification.
Figure 6 presents top‐view (top) and cross‐sectional (bottom) SEM images of CM LSA‐CB_PVdF_ electrodes subjected to different densification conditions: pristine, 3, 5, and 10 tons. The SEM images show that even the pristine electrodes, prepared using PVdF–NMP, exhibit a homogeneous morphology, forming a porous electronically conducting carbon network as presented in Figure 6a–d. This uniform structure is due to the solubility of PVMPT in NMP, which also explains the absence of PVMPT particles. As the applied pressure increases, no delamination of the electrode film from the current collector is observed (Figure 6g,h). However, a gradual reduction in porosity is noticeable as the pressure increases to 10 tons.
(a–d) Top‐view (top) and (e–h) cross‐sectional SEM images of CM LSA‐CBPVdF electrodes prepared under different densification conditions.
Figure 7a–f presents top‐view and cross‐sectional SEM images of CM LSA‐CB_CMC_ electrodes subjected to various densifying conditions. The pristine CMC‐based electrode surface appears noticeably non‐uniform (Figure 7a). Densification at 3 tons yields a more homogeneous morphology (Figure 7b), and increasing the pressure to 5 and 10 tons further enhances uniformity while visibly reducing porosity (Figure 7c,d). Cross‐sectional images reveal a porous conductive network formed by the additive around the polymer, particularly pronounced at 3 tons (Figure 7f). At 5 and 10 tons, however, delamination and particle cracking occur, indicating that excessive pressure causes contact loss, polymer damage, and further porosity reduction (Figure 7g,h). Similar trends occur when SBR is introduced as a co‐binder in different weight ratios (Table 1) and are likewise observed in other aqueous‐processed electrodes (Figures S6 and S7).
(a–d) Top‐view (top) and (e−h) cross‐sectional SEM images of CMC‐based electrodes prepared under different densification conditions. The indicators highlight delamination and particle cracking as the applied pressure increases to 5 and 10 tons.
This systematic pressure study demonstrated the balance between mechanical densification and electrode integrity across different binder systems.
Electrochemical Performance
4.2.2.3
Figure 8 compares the long‐term cycling stability across binder systems and applied pressures. PVdF binder system exhibits a trend where the discharge capacity starts high, decreases, and then increases again. This behavior is linked to electrolyte anion insertion, which slows down kinetics and structural changes in the polymer [15, 16]. A similar mechanism is observed in all aqueous processed electrodes, indicating that the (dis)charge process remains consistent regardless of the binder used. Furthermore, this trend is maintained across electrodes subjected to different levels of applied pressure, from pristine to 10‐ton densified electrodes.
Electrochemical performance of the investigated binder systems under various densifying conditions, evaluated at 1C rate over 1000 cycles (a) PVdF, (b) CMC, (c) CS55, and (d) CS73.
Figure 8a illustrates the electrochemical performance of PVdF‐based electrodes under different densification conditions. The pristine and 3‐ton densified electrodes exhibit higher maximum discharge capacities of 100 mAh g^−1^ compared to those densified at 5 and 10 tons, corresponds to 90% of the theoretical capacity. The electrodes densified at 10 t show a discharge capacity of 88 mAh g^−1^ over 100 cycles. Despite the reduced porosity at higher pressures, PVdF‐based electrodes maintain decent electrochemical performance.
Figure 8b–d illustrates the electrochemical performance of aqueous‐processed electrodes under different densification conditions, with the long‐term cycling stability of CMC‐based electrodes shown in Figure 8b. The pristine and 3‐ton densified CMC electrodes achieve specific discharge capacities of 101 and 104 mAh g^−1^, corresponding to 90% and 92% of the theoretical capacity. However, as pressure increases, electrochemical performance declines, with discharge capacities of 70 and 30 mAh g^−1^ at 5 and 10 tons, respectively. This decline can be attributed to electrode film delamination at higher pressure, polymer particle cracking, and reduced porosity, as observed in SEM images (Figure 7g,h).
The electrochemical performance of the electrodes processed with CMC and SBR in different weight ratios (refer to Table 1) was further investigated. Figure 8c displays the electrochemical performance of CS55‐based electrodes under different pressures. The pristine, 3, and 5 ton densified electrodes display similar electrochemical performance, achieving maximum discharge capacities of 93, 95, and 92 mAh g^−1^, corresponding to 83%, 86%, and 82% of the theoretical capacity, respectively. Herein, the addition of 50 wt% SBR enhances mechanical integrity and flexibility of the electrodes, allowing them to withstand higher pressure, as evidenced by the electrochemical observations. However, increasing the pressure to 10 tons reduces the specific discharge capacity to 40 mAh g^−1^ due to film delamination, reduced porosity, and particle cracking, as seen in Figure S6. Despite these issues, the electrochemical performance of 5‐ton densified CS55 electrodes surpasses that of both CMC‐ and PVdF‐based electrodes densified at 5 tons. Figure 8d depicts the electrochemical performance of CS73‐based electrodes (30 wt% SBR) at different pressures. The pristine, 3, and 5 ton densified CS73 electrodes exhibit similar electrochemical behavior, with discharge capacities of 75, 80, and 74 mAh g^−1^, corresponding to 67%, 72%, and 66% of the theoretical capacity, respectively. However, when the pressure is increased to 10 tons, the discharge capacity drops to 50 mAh g^−1^. Electrode film delamination particle cracking and reduced porosity, as observed in Figure S7, contribute to this decrease.
In summary, CMC‐based electrodes demonstrate electrochemical performance comparable to PVdF‐based systems under both pristine and 3‐ton densified conditions. However, at elevated pressures, delamination, reduced porosity, and particle cracking become evident in all aqueous‐processed electrodes. The addition of SBR enhances mechanical strength, enabling CS55 electrodes to perform better under moderate pressures. Conversely, as SBR content decreases, both electrode stability and electrochemical performance deteriorate. Overall, binder composition and densification conditions are critical factors in optimizing electrode performance. Figure 9 summarizes the parameters investigated and the corresponding conclusions, while the lower image highlights the synergy between the conductive additive choice, mixing method, and densification conditions.
(Top) Overview of the parameters investigated and the key take away highlighted. (Bottom) Synergistic effect of low surface area carbon black, low energy mixing, and densification.
Conclusion
5
This study systematically investigates the aqueous processing of PVMPT electrodes and examines the effects of conductive additive selection, mixing methods, and densification pressure on electrode microstructure and electrochemical performance. Regarding conductive additive selection, the use of a lower surface area carbon black (59 m^2^ g^−1^) minimized agglomeration and improved electrode homogeneity compared to the higher surface area carbon black (988 m^2^ g^−1^), which led to increased agglomeration. PVdF‐based electrodes exhibited good uniformity due to the solubility of PVMPT in NMP, whereas aqueous electrodes displayed an inhomogeneous structure. Attempts to enhance homogeneity through HEM method and hot‐pressing showed no notable improvements. While HEM method improved electrode uniformity, electrochemical performance remained suboptimal, likely because the intense collision of the balls mechanically damaged the PVMPT particles. Furthermore, the impact of densification pressure on electrode microstructure and electrochemical performance was explored. SEM analysis revealed that increasing pressure not only reduced the electrode porosity but also led to electrode film delamination from the current collector and the formation of microcracks in PVMPT. Electrodes produced with CMC and SBR exhibited different mechanical and electrochemical behaviors depending on their weight ratio. The CS55 formulation (50% CMC, 50% SBR) maintained better structural integrity under higher pressures, whereas decreasing SBR content led to a decline in mechanical strength and electrochemical performance. In summary, PVdF‐based electrodes demonstrated the best overall electrode microstructure and electrochemical performance, regardless of conductive additive type or densification pressure. Among aqueous binders, CMC‐based electrodes performed comparable to PVdF at lower pressures but demonstrated performance degradation at higher pressures. The addition of SBR improved mechanical strength, allowing CS55 electrodes to maintain better performance. Ultimately, binder composition, conductive additive selection, and densification pressure remain critical factors for optimizing the aqueous processing of PVMPT electrodes and enhancing electrochemical performance.
Supporting Information
Additional supporting information can be found online in the Supporting Information section. Supporting Fig. S1: (a, b) FT‐IR and Raman spectra of pristine PVMPT powder. Supporting Fig. S2: Surface SEM images of CM HSA‐CB and CM LSA‐CB pristine electrodes (non‐densified), prepared using PVdF and CMC binder. (a) CM HSA‐CB_PVdF_, (b) CM HSA‐CB_CMC_, (c) CM LSA‐CB_PVdF_, (d) CM LSA‐CB_CMC_. Supporting Fig. S3: Surface SEM images of HEM LSA‐CB_CMC_ electrodes fabricated using three different procedure and the corresponding electrochemical performance at a 1C rate (a–c) HEM 1 (d–f) HEM 2, and (g–i) HEM 3, respectively. Supporting Fig. S4: Surface SEM images of HSA‐CB electrodes processed via CM and HEM method. (a–b) CM HSA‐CB_CMC_ and (c–d) HEM 2 HSA‐CB_CMC_. Panel (e) shows the long‐term cycling stability of HEM 2 HSA‐CB_CMC_ at a 1C rate. Supporting Fig. S5: Top‐view SEM images of CM HSA‐CB_Binder_ electrodes densified under different conditions. Panels (a–c) show CM HSA‐CB_CMC_ electrodes (d–f) display CM HSA‐CB_CS55_ electrodes, while (g–i) present CM HSA‐CB_CS73_ electrodes subjected to different densification conditions: RT densification at 3 t, and hot‐pressing at 50 and 70°C with 1 t of pressure. Supporting Fig. S6: (a–d) Top‐view (top) and (e–h) cross‐sectional SEM images of CS55‐based electrodes prepared under different densification conditions. The indicators highlight delamination and particle cracking as the applied pressure increases to 5 and 10 t. Supporting Fig. S7: (a–d) Top‐view (top) and (e–h) cross‐sectional SEM images of CS73‐based electrodes prepared under different densification conditions. The indicators highlight delamination and particle cracking as the applied pressure increases to 5 and 10 t. Supporting Table S1: Overview of the abbreviations used in the manuscript and supporting information.
Funding
This study was supported by Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein‐Westfalen.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1D. Bresser , D. Buchholz , A. Moretti , A. Varzi , and S. Passerini , “Alternative Binders for Sustainable Electrochemical Energy Storage – the Transition To Aqueous Electrode Processing and Bio‐Derived Polymers,” Energy & Environmental Science 11, no. 11 (2018): 3096–3127.
- 2J. Li , J. Fleetwood , W. B. Hawley , and W. Kays , “From Materials to Cell: State‐of‐the‐Art and Prospective Technologies for Lithium‐Ion Battery Electrode Processing,” Chemical Reviews 122, no. 1 (2022): 903–956.34705441 10.1021/acs.chemrev.1c 00565 · doi ↗ · pubmed ↗
- 3N. Lingappan , L. Kong , and M. Pecht , “The Significance of Aqueous Binders in Lithium‐Ion Batteries,” Renewable & Sustainable Energy Reviews 147 (2021): 111227.
- 4A. M. Pillai , P. S. Salini , B. John , and M. T. Devassy , “Aqueous Binders for Cathodes: A Lodestar for Greener Lithium Ion Cells,” Energy & Fuels 36, no. 10 (2022): 5063–5087.
- 5R. Wang , L. Feng , W. Yang , et al., “Effect of Different Binders on the Electrochemical Performance of Metal Oxide Anode for Lithium‐Ion Batteries,” Nanoscale Research Letters 12, no. 1 (2017): 575.29086045 10.1186/s 11671-017-2348-6PMC 5662525 · doi ↗ · pubmed ↗
- 6L. Ibing , T. Gallasch , V. Göken , P. Niehoff , M. Winter , and M. Börner , “Making Aqueously Processed Li Ni 0.5 Mn 0.3Co 0.2O 2 ‐Based Electrodes Competitive in Performance: Tailoring Distribution and Interconnection of Active and Inactive Electrode Materials through Paste Surfactants,” ACS Applied Energy Materials 5, no. 11 (2022): 13155–13160.
- 7B. Esser , F. Dolhem , M. Becuwe , P. Poizot , A. Vlad , and D. Brandell , “A Perspective on Organic Electrode Materials and Technologies for Next Generation Batteries,” Journal of Power Sources 482 (2021): 228814.
- 8P. Poizot , J. Gaubicher , S. Renault , L. Dubois , Y. Liang , and Y. Yao , “Opportunities and Challenges for Organic Electrodes in Electrochemical Energy Storage,” Chemical Reviews 120, no. 14 (2020): 6490–6557.32207919 10.1021/acs.chemrev.9b 00482 · doi ↗ · pubmed ↗