A Complex Case of Ornithine Transcarbamylase Deficiency in a Patient With Severe Comorbid Conditions
Mobeen Khan, Sardar Ahmad Rafique, Suleman Khan, Asim Shah, Angraj Karan, Aizaz Anwar Khalid, Abdullah Afridi, Heela Tamim, Md Rubaiyat Tasfin Talukder

TL;DR
A 4-month-old boy with severe symptoms was diagnosed with ornithine transcarbamylase deficiency, a rare metabolic disorder, and stabilized with timely treatment.
Contribution
This case highlights the importance of early ammonia testing and nitrogen-scavenging therapy in diagnosing and managing partial OTCD in resource-limited settings.
Findings
Elevated ammonia and low citrulline levels indicated partial OTCD in a symptomatic infant.
Nitrogen-scavenging therapy and dietary adjustments stabilized the patient without dialysis.
Early diagnosis and treatment can prevent severe complications and improve outcomes in OTCD.
Abstract
Ornithine transcarbamylase deficiency (OTCD) is the most common urea‐cycle disorder, but it is often overlooked in Pakistan. It can present after the neonatal period with symptoms resembling sepsis and significant respiratory alkalosis. We report a 4‐month‐old boy with fever, lethargy, poor feeding, and loose stools for 5 days. He arrived with a rapid heart rate and quick breathing. His arterial blood gas showed a pH of 7.65, pCO2 of 14 mmHg, and HCO3 − of 15.5 mmol/L. Plasma ammonia levels were elevated at 78.1 μmol/L (reference range 11–32). A quantitative amino acid test revealed greatly increased glutamate (263 μmol/L; reference range 18–98) with low citrulline (6 μmol/L; reference range 16–32) and a normal ornithine/arginine ratio, which matches the pattern seen in partial OTCD. A contrast‐enhanced CT scan of the brain showed widespread leptomeningeal enhancement and mild…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
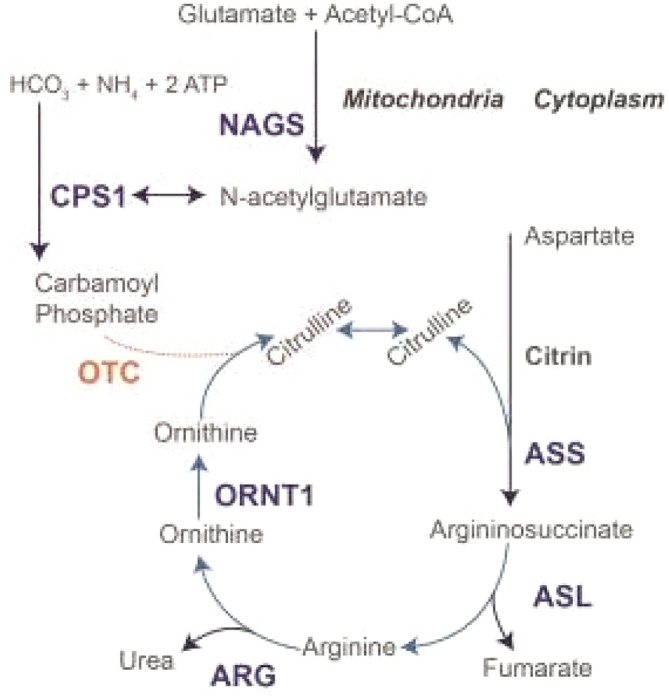 FIGURE 1
FIGURE 1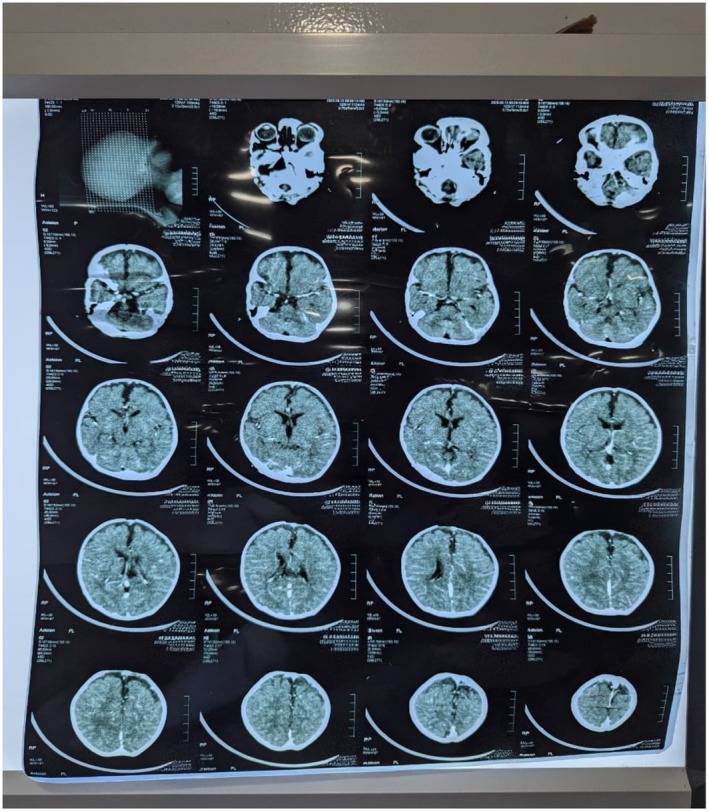 FIGURE 2
FIGURE 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism and Genetic Disorders · Folate and B Vitamins Research · Alcoholism and Thiamine Deficiency
Introduction
1
Ornithine transcarbamylase deficiency is a rare X‐linked disorder of the urea cycle, with an incidence of 1 in 50,000 to 80,000. It was first reported in 1962 by Russell in two girls, aged 20 months and 6 years, who were found to have hyperammonemia associated with episodic vomiting, delirium, stupor, failure to thrive, and mental retardation [1].
The urea cycle involves six enzymes that remove nitrogen from the body by converting neurotoxic ammonia into urea. One of these enzymes is Ornithine Transcarbamylase (OTC). A deficiency of this enzyme results in the excessive accumulation of ammonia, which is a potent neurotoxin, as shown in Figure 1 [2].
Urea cycle depicting the six enzymes involved and how OTC deficiency causes Ornithine buildup and decreases Citrulline production [2].
Ornithine transcarbamylase (OTC) deficiency can manifest as a severe neonatal onset disease in males and as a post‐neonatal onset disease (partial deficiency) in both males and females [2]. The infant becomes symptomatic after feeding starts because human milk supplies a protein load that is converted to ammonia. Severe neonatal onset Ornithine transcarbamylase deficiency typically appears on days two to three of life and is usually life‐threatening by the time medical attention is sought [3]. Symptomatic carriers often have a lifelong history of migraines, recurrent vomiting, abnormal behavior characterized by disorientation and confusion, anorexia, and avoidance of high‐protein foods [4].
Ornithine transcarbamylase deficiency (OTCD), although the most common urea cycle disorder worldwide, remains an exceptionally rare condition in Pakistan, with its true incidence unknown due to lack of newborn screening programs and underreporting; global estimates suggest a prevalence of 1 in 56,000–80,000 live births. In resource‐limited settings like Pakistan, delayed recognition and limited access to specialized metabolic diagnostics further increase morbidity and mortality. Therefore, documenting individual OTCD cases is crucial to raise clinical awareness, improve understanding of its presentation in the Pakistani population, and highlight the urgent need for early diagnostic strategies and multidisciplinary management to enhance patient outcomes.
Case History
2
A 4‐month‐old boy was born at 38 weeks via normal vaginal delivery to a mother with four prior pregnancies. He weighed 3.2 kg at birth. At 5 days old, he presented with fever, lethargy, poor oral intake, and loose stools. He had a delayed cry at birth and was admitted as a newborn for suspected sepsis. Upon arrival, he appeared pale and malnourished. He was tachycardic and tachypneic, with significant lethargy and poor feeding. His pulses were strong, the precordium was soft, and his heart sounds were normal.
Differential Diagnosis
2.1
Based on his presentation of lethargy, poor feeding, tachypnea, and a history suggestive of neonatal sepsis, the initial considerations included:
- Sepsis
- Inborn errors of metabolism (e.g., urea cycle disorders)
- Electrolyte disturbances
- Neurological causes (intracranial infection, structural abnormality)
Investigations
2.2
Serum urea was elevated at 77.4 mg/dL, while creatinine remained normal. Liver function tests were slightly abnormal during the acute illness but normalized before discharge. Arterial blood gas revealed a pH of 7.65, pCO_2_ of 14 mmHg, and HCO_3_ ^−^ of 15.5 mmol/L, indicating primary respiratory alkalosis with partial metabolic compensation. Plasma ammonia was 78.1 μmol/L (reference 11–32), indicating hyperammonemia.
A quantitative plasma amino acid profile showed elevated glutamate at 263 μmol/L (reference 18–98) and decreased citrulline at 6 μmol/L (reference 16–32). Arginine and ornithine were normal. This biochemical profile, together with hyperammonemia and respiratory alkalosis, strongly suggested ornithine transcarbamylase deficiency (OTCD). PTH‐intact was 32.66 pg/mL, within the normal range.
Contrast‐enhanced CT brain imaging revealed diffuse leptomeningeal enhancement with mild dilation of the bifronto‐temporal subarachnoid spaces (BESS pattern), with no focal lesions, mass effects, midline shifts, or hydrocephalus (Figure 2). Echocardiography showed a small ASD and a tiny PDA with left‐to‐right shunts, balanced chambers, good biventricular function, and normal cardiac situs. A single right SVC with a left innominate vein was noted. Outpatient cardiology follow‐up was advised in 6 months.
CT scan of the brain showing diffuse leptomeningeal enhancement and mild bifronto‐temporal subarachnoid space prominence (BESS pattern); no focal lesion or mass effect.
Treatment
2.3
The child was treated with broad‐spectrum antibiotics (meropenem and ceftriaxone/fortum) for suspected sepsis. Ammonia‐lowering therapy was initiated with sodium benzoate. Additional supportive treatments included L‐arginine, L‐carnitine, folic acid, zinc, and calcium. His feeding plan was adjusted to a protein‐restricted, formula‐based diet (Basic‐P/Morinaga BF‐1).
Conclusion and Results
3
The infant showed gradual clinical improvement with better feeding and a steady decline in ammonia levels. Liver tests normalized before discharge. Outpatient follow‐up was recommended, and the child was discharged in a stable condition.
Discussion
4
This case highlights several key points about ornithine transcarbamylase deficiency (OTCD). First, it demonstrates that partial or proximal urea‐cycle defects can present beyond the neonatal period with vague symptoms such as lethargy, poor feeding, and gastrointestinal upset, along with respiratory alkalosis that exceeds what the overall illness would suggest. These signs should trigger testing for ammonia levels, even if sepsis appears likely. The combination of high ammonia, low plasma citrulline, and normal levels of ornithine and arginine strongly indicates OTCD, which allowed for prompt treatment in this case. Including this reflex in pediatric emergency protocols is essential, especially in resource‐limited settings where delayed diagnosis can greatly worsen outcomes. International guidelines also emphasize that even mild hyperammonemia can damage the brain. This underscores the importance of rapid diagnosis and ammonia management, regardless of the peak value [5, 6, 7, 8, 9].
Second, the infant's imaging offers valuable teaching points. While CT suggests leptomeningeal enhancement and benign enlargement of the bifrontotemporal subarachnoid spaces (BESS), MRI with MR spectroscopy (MRS) is the most informative imaging method for urea cycle disorders. MRS can identify the characteristic increase in brain glutamine (along with decreased myo‐inositol), even if structural scans look normal. Changes seen on diffusion‐weighted imaging often involve the insular and peri‐insular cortex, the occipital lobes, the basal ganglia, and the dorsal thalamus during acute injury. These patterns are linked to neurocognitive issues in executive functions. When available, MRS helps distinguish metabolic encephalopathy from infectious meningitis when CT results are unclear [10, 11].
Third, management in this case follows established pathways. It involves quickly reducing ammonia levels by (i) temporarily stopping external protein intake and increasing calories to reverse catabolism, (ii) using nitrogen‐scavengers like sodium benzoate, phenylacetate, or oral phenylbutyrate, and (iii) administering urea‐cycle intermediates such as arginine. Data from an extensive multicenter study show improved survival with phenylacetate or benzoate during hyperammonemic crises. Dialysis is reserved for severe or refractory hyperammonemia, based on pediatric kidney replacement guidelines. The infant's relatively moderate peak ammonia allowed for a medical and dietary approach without dialysis, in line with existing algorithms [8, 9, 12, 13].
Fourth, having congenital heart disease (secundum ASD, tiny PDA) along with temporary kidney problems is a practical concern. Cardiorespiratory issues can mask the signs of respiratory alkalosis, and reduced kidney function can affect how the body clears benzoate and manages fluids during acute care. These concerns are specifically mentioned in guidelines that recommend close monitoring of heart and kidney function during therapy [5, 9, 13].
Ultimately, this case from a setting with limited screening underscores the importance of confirming the diagnosis through molecular testing and offering family counseling. Resources like GeneReviews emphasize that female relatives may be symptomatic carriers. Cascade testing, along with sick‐leave policies and emergency procedures, can help reduce the risk of future crises. Meanwhile, closely managing protein intake, reintroducing it once ammonia levels normalize, regularly monitoring amino acids, and having plans for early escalation are essential for safety in outpatient care [6, 7, 8, 9, 14].
Author Contributions
Mobeen Khan: conceptualization, methodology, project administration, supervision. Sardar Ahmad Rafique: visualization, writing – original draft, writing – review and editing. Suleman Khan: conceptualization, methodology, writing – original draft, writing – review and editing. Asim Shah: validation, visualization, writing – original draft. Angraj Karan: writing – original draft, writing – review and editing. Aizaz Anwar Khalid: methodology, resources. Abdullah Afridi: methodology. Heela Tamim: methodology, writing – original draft. Md Rubaiyat Tasfin Talukder: visualization, writing – review and editing.
Funding
The authors have nothing to report.
Ethics Statement
The authors have nothing to report.
Consent
Written informed consent was obtained from the patient and/or their legal guardian for the publication of this case report, including relevant clinical details and any accompanying images.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A. G. Campbell , L. E. Rosenberg , P. J. Snodgrass , and C. T. Nuzum , “Ornithine Transcarbamylase Deficiency: A Cause of Lethal Neonatal Hyperammonemia in Males,” New England Journal of Medicine 288, no. 1 (1973): 1–6, 10.1056/NEJM 197301042880101.4681895 · doi ↗ · pubmed ↗
- 2J. H. Choi , B. H. Lee , J. H. Kim , et al., “Clinical Outcomes and the Mutation Spectrum of the OTC Gene in Patients With Ornithine Transcarbamylase Deficiency,” Journal of Human Genetics 60, no. 9 (2015): 501–507, 10.1038/jhg.2015.54.25994866 · doi ↗ · pubmed ↗
- 3N. E. Maestri , D. Clissold , and S. W. Brusilow , “Neonatal Onset Ornithine Transcarbamylase Deficiency: A Retrospective Analysis,” Journal of Pediatrics 134, no. 3 (1999): 268–272, 10.1016/s 0022-3476(99)70448-8.10064660 · doi ↗ · pubmed ↗
- 4H. Mendez‐Figueroa , K. Lamance , V. R. Sutton , K. Aagaard‐Tillery , and I. van den Veyver , “Management of Ornithine Transcarbamylase Deficiency in Pregnancy,” American Journal of Perinatology 27, no. 10 (2010): 775–784, 10.1055/s-0030-1254240.20458665 · doi ↗ · pubmed ↗
- 5J. Häberle , A. Burlina , A. Chakrapani , et al., “Suggested Guidelines for the Diagnosis and Management of Urea Cycle Disorders: First Revision,” Journal of Inherited Metabolic Disease 42, no. 6 (2019): 1192–1230, 10.1002/jimd.12100.30982989 · doi ↗ · pubmed ↗
- 6U. Lichter‐Konecki , L. Caldovic , H. Morizono , and M. Tuchman , “Ornithine Transcarbamylase Deficiency,” in Gene Reviews, ed. M. P. Adam , S. Bick , G. M. Mirzaa , R. A. Pagon , S. E. Wallace , and A. Amemiya (University of Washington, 2022).24006547 · pubmed ↗
- 7S. Matsumoto , J. Häberle , J. Kido , H. Mitsubuchi , F. Endo , and K. Nakamura , “Urea Cycle Disorders—Update,” Journal of Human Genetics 64, no. 9 (2019): 833–847, 10.1038/s 10038-019-0614-4.31110235 · doi ↗ · pubmed ↗
- 8N. Savy , D. Brossier , C. Brunel‐Guitton , L. Ducharme‐Crevier , G. Du Pont‐Thibodeau , and P. Jouvet , “Acute Pediatric Hyperammonemia: Current Diagnosis and Management Strategies,” Hepatic Medicine: Evidence and Research 10 (2018): 105–115, 10.2147/HMER.S 140711.30254497 PMC 6140721 · doi ↗ · pubmed ↗