The risk of radiation necrosis from combined radiotherapy and BRAF inhibitor in lung adenocarcinoma brain metastases: a comprehensive review and future perspectives
Lina Yang, Yanan He, Yong Peng, Mao Sun, Zheng Tang, Liang Du, Yongzhong Wu, Wei Zhou, Dingyi Yang

TL;DR
This paper reviews how combining BRAF inhibitors with radiotherapy increases the risk of brain tissue damage in lung cancer patients with brain metastases and suggests ways to reduce this risk.
Contribution
The paper provides a comprehensive analysis of the mechanisms and risk factors for radiation necrosis in BRAF inhibitor and radiotherapy combination therapy for lung adenocarcinoma brain metastases.
Findings
BRAF inhibitors exacerbate radiotherapy-induced vascular injury and blood-brain barrier disruption.
Combination therapy increases inflammatory responses and risks of radiation necrosis through MAPK and VEGF pathway modulation.
Strategies like optimized treatment sequencing and advanced imaging are proposed to mitigate radiation necrosis risks.
Abstract
The management of lung adenocarcinoma with brain metastases (BMs) is particularly challenging when BRAF-V600E mutations emerge as a resistance mechanism to EGFR tyrosine kinase inhibitors. While the combination of BRAF/MEK inhibitors (e.g., dabrafenib and trametinib) and radiotherapy (RT) is a pivotal therapeutic strategy, it significantly increases the risk of radiation necrosis (RN). This review summarizes the current understanding of the molecular mechanisms and risk factors underlying RN development in this specific patient population. We detail how BRAFi exacerbate RT-induced vascular injury, blood-brain barrier (BBB) disruption, and inflammatory responses, focusing on MAPK pathway modulation, VEGF signaling inhibition, and paradoxical pathway activation. Clinical correlations regarding treatment timing and regimen choice are discussed. Finally, we propose comprehensive strategies…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
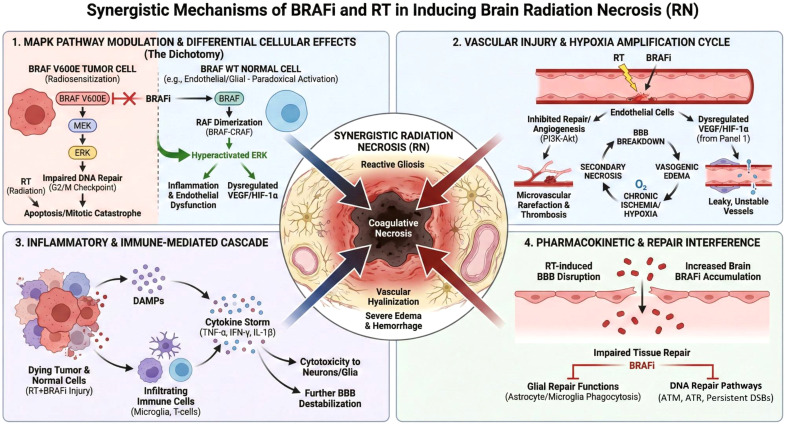 Figure 1
Figure 1| Inhibitor (Trade name) | Generic name | Primary target specificity | Key approved indication(s) | Notable clinical trial (e.g.) |
|---|---|---|---|---|
| Zelboraf | Vemurafenib | Selective for BRAF V600 mutant | Unresectable or metastatic melanoma with BRAF V600E mutation | BRIM-3 ( |
| Tafinlar | Dabrafenib | Selective for BRAF V600 mutant | Melanoma, NSCLC, ATC with BRAF V600E mutation | BREAK-3 ( |
| Braftovi | Encorafenib | Selective for BRAF V600 mutant | Used with binimetinib/cetuximab for BRAF-mutant melanoma/CRC | COLUMBUS; BEACON CRC ( |
| Study | Study year | Study type | N (patients) | Median dose (Gy) | Radiotherapy | Targeted therapy | Toxicity incidence/description | Start of targeted therapy | Tumor type | Subgroup analysis |
|---|---|---|---|---|---|---|---|---|---|---|
| Kim et al. ( | 2017 | Retrospective cohort study | 218 (concurrent TTs) | 24 (18–24) | SRS± WBRT | VEGFR, EGFR, ALK, BRAF inhibitors | Brain necrosis: 8.8–15.6%; VEGFR TKIs increased RN risk (13% vs. 5.3%); EGFR TKIs elevated RN risk (14% vs. 5.3%). BRAF-i showed no significant increase in RN. | Concurrent | BMs (NSCLC, renal, melanoma, and breast) | EGFR TKIs with SRS+WBRT significantly elevated RN risk (20% vs. 3.3%). Peaking 4.5–9 months post-SRS with concurrent therapies |
| Kroeze et al. ( | 2017 | Systematic Review | 644 (cranial SRT) | 18–27 | SRT | BRAF, EGFR, ALK inhibitors | BRAFi: Grade 3/4 cerebral oedema in 15%; intratumoral hemorrhage in up to 23%. | Concurrent | BMs, melanoma, NSCLC | Higher risk of severe CNS toxicity with BRAF-i. But EGFR-i show good tolerability with cranial SRT. |
| Pierre-Yves et al. ( | 2021 | Systematic Review | 327 patients | 24 (6–24), 21–24, 25 | SRT | Trastuzumab and T-DM1, EGFR, BRAF, MEK, ALK inhibitors | No significant increase in RN or clinical toxicity | Concurrent or at the time of SRT (± 30 days) | BMs (melanoma, lung, breast, renal carcinoma) | Highlighted safety of concurrent SRT with TTs, with no clinically significant increase in RN |
| Maikel et al. ( | 2017 | Literature Review | Multiple studies | 20–30 (WBRT); 15–20 (SRS) | WBRT, SRS | EGFR, BRAFi, Sunitinib, Sorafenib | Erlotinib, gefitinib and vemurafenib showed increased neurotoxicity risks (fatigue, brain hemorrhage). | Concurrent | BMs | Most TTs don’t increase neurotoxicity with CRT. But sunitinib and Sorafenib may act as radiosensitizers. |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBrain Metastases and Treatment · Melanoma and MAPK Pathways · Lung Cancer Treatments and Mutations
Introduction
1
Lung adenocarcinoma, the predominant subtype of non-small cell lung cancer (NSCLC), is frequently driven by actionable oncogenic mutations. While EGFR mutations are effectively targeted by tyrosine kinase inhibitors (TKIs) such as osimertinib, acquired resistance often develops. The BRAF-V600E mutation has emerged as a key resistance mechanism, shifting the treatment paradigm to alternative targeted therapies (TTs), including dabrafenib and trametinib (1, 2). Brain metastases (BMs) develop in approximately 20-40% of advanced NSCLC patients and are a leading cause of morbidity and mortality, with a median overall survival of less than one year (3, 4). Local radiotherapy (RT), including stereotactic radiosurgery (SRS) and whole-brain radiotherapy (WBRT), remains a cornerstone of BMs management, providing durable local control (5–7). Combining RT with targeted agents has shown promise for improving intracranial control, yet the optimal integration of these modalities remains undefined, particularly for BRAF inhibitors (8, 9).
Complicating this approach is the risk of RN, a serious complication of cranial irradiation characterized by neuroinflammation, blood-brain barrier disruption, and cognitive decline (10–13). The combination of BRAF inhibitors (BRAFi) with radiotherapy (RT) has shown efficacy in controlling BRAF-mutant tumors but is associated with an increased risk of severe radiation necrosis, particularly in the central nervous system (CNS) (14). When BRAFi (especially first-generation inhibitors like vemurafenib) are administered concurrently with RT, they may synergistically disrupt critical pro-survival and repair pathways in the vascular endothelium. Furthermore, the “paradoxical activation” of the MAPK pathway by these inhibitors in cells with wild-type BRAF (such as normal endothelial cells in the irradiated field) may create a pro-inflammatory milieu, enhancing vascular leakage and inflammatory cell recruitment. This combined insult amplifies the cascade of endothelial dysfunction, abnormal angiogenesis, and eventual vessel obliteration, thereby significantly increasing the probability of developing severe, symptomatic RN (15, 16).
This review synthesizes current evidence on the mechanisms and risk factors for RN in patients with BRAF-mutant lung adenocarcinoma receiving combined BRAFi and RT. We highlight the urgent need for multidisciplinary management strategies and future research to optimize therapeutic outcomes while mitigating this debilitating toxicity.
BRAF inhibitors: classification and mechanism of action
2
BRAFi are a class of targeted anticancer drugs designed to block the activity of the BRAF kinase, a key component of the MAPK/ERK signaling pathway. This pathway is frequently dysregulated in cancers, most commonly via the BRAF V600E mutation. BRAFi specifically bind to the ATP-binding site of the mutant BRAF kinase, inhibiting its constitutive activity and downstream signaling, leading to cell cycle arrest and apoptosis (17).
It is crucial to distinguish their mode of action regarding wild-type BRAF (BRAF WT). First-generation BRAFi (e.g., vemurafenib, dabrafenib) are considered selective or mutant-selective inhibitors. They potently inhibit monomeric mutant BRAF (especially V600E). However, in cells with active upstream signaling (e.g., RAS activation), they can paradoxically activate the MAPK pathway by promoting dimerization of BRAF WT with CRAF, a phenomenon known as “paradoxical activation” (18). Second-generation “paradox breaker” inhibitors (e.g., PLX8394) are designed to inhibit both mutant and wild-type BRAF without inducing this paradoxical activation (19). Pan-RAF inhibitors (e.g., sorafenib, LY3009120) target all RAF isoforms (ARAF, BRAF, CRAF) regardless of mutation status (20, 21). Table 1 lists the current available BRAFi in market.
Overview of radiation-induced tissue injury, radiation necrosis, and vascular remodeling
3
Radiation-Induced Tissue Injury (RITI) is a broader term encompassing the pathological changes in normal tissues following radiotherapy. It is a dynamic process involving acute inflammation and chronic, progressive fibrosis. The pathogenesis involves DNA damage in parenchymal and vascular endothelial cells, triggering a persistent pro-inflammatory and pro-fibrotic cytokine cascade (22).Radiation necrosis(RN) is a delayed, non-malignant complication of CNS by RT, typically occurring 3 months to several years post-treatment, characterized by focal coagulative necrosis of the white matter. It results from a combination of direct glial cell damage and, crucially, vascular endothelial injury, leading to increased blood-brain barrier(BBB) permeability, edema, and thrombosis (23).
RN primarily driven by vascular dysfunction, immune-mediated inflammation, and oxidative stress, all contributing to disruption of the BBB, ischemic injury, and chronic neuroinflammation (24). In which, initiated primarily by radiotherapy-induced disruption of BBB and direct neural cell damage. Ionizing radiation causes structural and functional damage to the BBB by inducing endothelial cell apoptosis, tight junction protein degradation, and increased vascular permeability, leading to leakage of plasma components and infiltration of inflammatory cells into the brain parenchyma (25). This BBB disruption facilitates neuroinflammation and oxidative stress, which are pivotal in the pathogenesis of RN (24). Key molecular pathways involved include VEGF-mediated endothelial damage, cGAS-STING-driven innate immune activation, and radiation-induced mitochondrial dysfunction (13).
Microglial activation is a hallmark of RN, with these resident immune cells releasing pro-inflammatory cytokines such as IL-6, TNF-α, and reactive oxygen species (ROS), exacerbating neuronal injury and perpetuating a vicious cycle of inflammation. Oxidative stress, resulting from excessive ROS production and impaired antioxidant defenses, further damages lipids, proteins, and nucleic acids within neural cells, contributing to cellular dysfunction and death (26, 27).
Several factors influence the risk of RN, including radiation dose, fractionation schedule, and treatment volume. Higher biologically effective doses (BED), larger treatment fields, and retreatment with cumulative doses exceeding 100 Gy increase RN risk (28). Additionally, tumor biology (e.g., BRAF mutations, VEGF overexpression) and patient-specific factors (e.g., age, pre-existing vascular disease) may further predispose individuals to RN (29).
(3) Radiation directly damages vascular endothelial cells, inducing senescence, apoptosis, and increased expression of adhesion molecules (e.g., ICAM-1, VCAM-1). This initiates a chronic state of vascular inflammation and dysfunction. Key alterations include: Loss of nitric oxide (NO) bioavailability, increased oxidative stress, and upregulation of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IFN-γ). Disruption of tight junctions and the basement membrane increases permeability. Radiation promotes the expression of VEGF and other angiogenic factors as a compensatory response. However, the newly formed vessels are often abnormal—dilated, tortuous, and leaky—with poor pericyte coverage, failing to restore proper perfusion. Chronic inflammation leads to vessel wall thickening, hyalinization, and thrombosis, culminating in reduced vascular density and tissue ischemia. The persistent release of TGF-β drives the differentiation of fibroblasts into myofibroblasts, resulting in perivascular and parenchymal fibrosis, which further compromises tissue architecture and function (30, 31).
Clinical evidence of RN risk with BRAF inhibitors and RT
4
The combination of RT with molecular targeted therapies (TTs) represents a cornerstone of modern oncology, offering potent synergy for tumor control. However, this synergy can also amplify off-target toxicity in normal tissues. In the context of brain metastases, concurrent RT and BRAFi has been increasingly associated with an elevated risk of RN (29, 32).
The clinical evidence supporting this association, however, is unevenly distributed across cancer types. As summarized in Table 2, the most compelling data originate from studies in melanoma and glioblastoma, where the use of BRAFi is well-established. These studies consistently report a higher incidence and sometimes atypical presentation of RN following SRS or fractionated RT when combined with targeted agents. For instance, a illustrative case involved a patient with MAP2K1-mutant melanoma who developed RN nine months post-SRS while on trametinib and low-dose dabrafenib, which was subsequently managed with bevacizumab (33). This and other cases underscore the clinical reality of this interaction.
In stark contrast, direct clinical evidence specifically linking BRAFi to RN in NSCLC remains scarce. The limited number of studies, as cataloged in Table 2, highlights a significant knowledge gap. This paucity of data is particularly concerning given the unique pathophysiology of lung cancer brain metastases and the increasing adoption of these targeted therapies in the NSCLC setting. The mechanisms underlying this increased risk—potentially involving radiosensitization, vascular toxicity, inflammatory and tissue repair interference—are explored in the following section.
Synergistic mechanisms of BRAFi and RT in inducing radiation necrosis
5
BRAFi, used particularly in BRAF-mutant melanoma and lung adenocarcinoma, may potentiate radiation-induced neurotoxicity through mechanisms that are not fully elucidated but likely involve exacerbation of oxidative stress and inflammatory responses (16, 32). BRAFi such as dabrafenib and MEK inhibitors such as trametinib target the MAPK/ERK signaling pathway, suppressing tumor proliferation. When combined with radiation, BRAFi may amplify additional toxicities by enhancing radiosensitivity through several mechanisms:
Radiosensitization via MAPK pathway modulation
5.1
The combination of BRAFi with RT leads to complex, cell-context-dependent interactions within the MAPK/ERK pathway that ultimately enhance radiation toxicity. In BRAF V600E-mutant tumor cells, BRAFi potently inhibit constitutive MAPK signaling. This inhibition not only suppresses proliferation but also critically impairs the cellular response to radiation-induced DNA damage. Specifically, BRAFi abrogate the radiation-induced G2/M cell-cycle checkpoint arrest, a vital period for DNA repair, forcing cells with unrepaired double-strand breaks into mitosis, leading to mitotic catastrophe and enhanced cell death. This constitutes a primary mechanism of radiosensitization in the tumor compartment (34).
Paradoxically, in BRAF wild-type (WT) cells—such as vascular endothelial cells, normal glial cells, and infiltrating immune cells within the tumor microenvironment—first-generation BRAFi can induce hyperactivation of the MAPK pathway. This occurs through drug-induced promotion of RAF dimerization (e.g., BRAF WT-CRAF), leading to feedback-driven, ligand-dependent ERK signaling. In the context of an irradiated field, this “paradoxical activation” in normal tissues exacerbates radiation-induced stress, promoting excessive inflammatory cytokine release, endothelial dysfunction, and cytotoxic effects on supportive glial cells. This differential effect—inhibition in tumor cells versus potential activation in surrounding normal tissue—creates a dichotomy that underlies both the therapeutic efficacy and the heightened risk of normal tissue complications like radiation necrosis (18, 35).
The phenomenon of “paradoxical activation” is central to the vascular toxicity of combined therapy. In normal cells with intact RAS signaling, BRAFi binding to one protomer of a RAF dimer (e.g., BRAF) allosterically promotes transactivation of the drug-free partner (e.g., CRAF). This leads to hyperactivation of the MEK-ERK pathway (17). In the irradiated tissue, which is already hypoxic and rich in growth factors, this paradoxically activated ERK signaling further stabilizes HIF-1α and increases VEGF production. However, this VEGF surge is pathologic. It promotes the formation of leaky, malformed, and unstable vessels that lack proper pericyte coverage and are incapable of adequate perfusion (36). The MAPK pathway is integral to VEGF-mediated angiogenic signaling, its inhibition by BRAFi disrupts the compensatory angiogenic response necessary for recovery from RT-induced damage (37). Figure 1 illustrates the possible synergistic mechanism of the combined use of BRAFi and RT in inducing radiation necrosis.
Synergistic mechanisms of BRAFi and RT in inducing radiation necrosis.
Vascular injury and hypoxia amplification
5.2
The synergistic vascular toxicity of BRAFi and RT is a cornerstone in the pathogenesis of severe RN. RT directly damages vascular endothelial cells, initiating a cascade of senescence, apoptosis, and increased permeability. BRAFi potentiate this injury by inhibiting endothelial cell proliferation, migration, and survival signals that are crucial for vascular repair and maintenance. The combined insult leads to microvascular rarefaction, characterized by a loss of functional capillaries, and persistent BBB breakdown.
BRAFi compromise endothelial repair, leading to increased vascular permeability, microvascular thrombosis, and sustained edema (16). VEGF upregulation, exacerbated by RT, promotes capillary leakage and inflammatory infiltration, further weakening the BBB and exacerbating radiation-induced injury (38). The failure to regenerate a competent vascular network results in chronic ischemia and hypoxia within the irradiated tissue bed. This hypoxic environment, in turn, promotes secondary necrosis, exacerbates edema, and creates a vicious cycle that drives the expansion of necrotic lesions. Histopathologically, this is evident as fibrinoid necrosis, vessel wall hyalinization, and thrombosis.
BRAFi increase ROS production and inhibit NRF2-mediated antioxidant defense, leading to prolonged oxidative stress and cellular injury. Radiation exposure already generates ROS, and the combined effect may amplify oxidative damage in both tumor and normal brain tissue (39, 40).
Inflammatory and immune-mediated mechanisms
5.3
The convergence of BRAFi and RT potently activates and sustains a pro-inflammatory milieu that amplifies tissue damage. BRAFi enhance antigen presentation and increase the release of damage-associated molecular patterns (DAMPs) from dying tumor cells. Concurrently, RT induces a robust sterile inflammatory response by releasing DAMPs from both tumor and injured normal cells. The combination leads to an amplified recruitment and activation of innate immune cells (e.g., microglia, macrophages) and adaptive immune cells (e.g., T-cells) into the tumor microenvironment and surrounding brain parenchyma (34, 41).
This infiltrate releases high levels of pro-inflammatory cytokines such as TNF-α, IFN-γ, and IL-1β. These cytokines further destabilize the BBB, promote vasogenic edema, and exert direct cytotoxic effects on oligodendrocytes and neurons. The resulting histopathology is a blend of coagulative necrosis, intense perivascular and parenchymal lymphocytic inflammation, and reactive gliosis—features that reflect the compounded effect of both treatments and distinguish this form of necrosis from that caused by RT alone.
Tissue repair and pharmacokinetic interference
5.4
Emerging evidence suggests that BRAFi may interfere with the intrinsic repair capacity of the CNS beyond direct vascular toxicity (16). Astrocytes and microglia, essential for maintaining homeostasis and coordinating repair after injury, rely on finely tuned intracellular signaling. BRAFi exposure can impair the protective and reparative functions of these glial cells, potentially by disrupting MAPK-dependent trophic support and phagocytic activity. This impairment slows the clearance of cellular debris and the resolution of inflammation, prolonging the injury phase (17, 30).
Furthermore, the RT-induced disruption of the BBB may alter the pharmacokinetics of BRAFi. The compromised barrier can lead to increased and sustained local drug accumulation in the irradiated brain tissue, resulting in higher-than-expected exposure and prolonged on-target (and off-target) effects on both tumor and normal cells. This localized pharmacokinetic shift creates a focus of intensified pharmacodynamic activity, concentrating the mechanisms described above (vascular injury, paradoxical signaling, inflammation) precisely within the region most vulnerable to necrosis, thereby explaining the focal and severe nature of this complication (16, 39, 42).
Mechanistic interplay between BRAFi and VEGF signaling in the context of RT
6
Direct suppression of tumor VEGF signaling
6.1
In BRAF V600E-mutant tumor cells, the constitutively active BRAF-MEK-ERK axis drives oncogenic gene expression programs, including the stabilization of Hypoxia-Inducible Factor 1-alpha (HIF-1α) and the transcription of pro-angiogenic factors like VEGF-A. BRAFi directly block this pathway, leading to decreased ERK-mediated phosphorylation events, reduced HIF-1α protein stabilization, and subsequent downregulation of VEGF transcription and secretion (15). This anti-angiogenic effect is therapeutically beneficial, as it reduces tumor neovascularization, normalizes the chaotic tumor vasculature, and can improve drug delivery. However, when combined with radiotherapy (RT), this systemic suppression of pro-repair VEGF signaling can be detrimental. In normal tissues within the radiation field, the impaired angiogenic response prevents the necessary vascular repair after RT-induced injury, exacerbating ischemia and culminating in necrosis (30).
Dual and paradoxical effects on VEGF signaling
6.2
The impact of BRAFi on VEGF signaling is profoundly context-dependent, creating a dichotomy between tumor and normal tissue. While BRAFi suppress the pathway in mutant cells, they can paradoxically enhance MAPK and VEGF signaling in BRAF WT cells, such as endothelial cells, fibroblasts, and astrocytes (17). This aberrant activation can lead to increased HIF-1α activity and VEGF secretion in the stromal compartment, resulting in a dysregulated, non-productive angiogenic stimulus.
Impaired angiogenic repair post-radiotherapy
6.3
RT inflicts direct damage on the microvasculature, causing endothelial cell apoptosis and capillary rupture. The natural tissue response to this injury is a compensatory upregulation of VEGF-driven angiogenesis to restore perfusion and initiate repair. BRAFi disrupt this critical recovery process. By suppressing VEGF signaling, they impair endothelial cell proliferation, migration, and survival, effectively blocking the regeneration of a functional vascular network (41). What would otherwise be a transient, repairable injury becomes a state of chronic ischemia, hypoxia, and ultimately, progressive necrosis.
Clinical risk factors, manifestations and diagnosis of RN
7
Clinical data, primarily from melanoma, confirm the heightened RN risk with BRAFi+RT combination. Key risk factors include: Treatment Timing: Concurrent or closely sequenced administration (within ~3 days) carries the highest risk (16). RT Modality and Dose: SRS, with its high dose per fraction, is associated with a greater risk of symptomatic RN compared to fractionated WBRT when combined with BRAFi, likely due to more intense focal vascular injury (43).
RN manifests clinically in distinct phases with characteristic symptoms and imaging features that evolve over time. In the acute phase, patients commonly present with symptoms such as headache, cognitive dysfunction, and neurological deficits, reflecting early inflammatory and edema processes in the brain tissue. These acute symptoms may be transient but are indicative of initial radiation-induced cellular and vascular stress. As the injury progresses to the delayed or late phase, more chronic manifestations emerge, including white matter abnormalities, brain atrophy, and persistent cognitive decline. These late effects correspond to irreversible structural damage such as demyelination, necrosis, and microvascular injury (44, 45).
Advanced neuroimaging techniques, such as magnetic resonance imaging (MRI), in which including conventional T2-weighted imaging and diffusion tensor imaging (DTI), reveals white matter changes, edema, and atrophic changes, play a pivotal role in the diagnosis and monitoring of RN. But, distinguishing RN from tumor progression is challenging, particularly in patients on targeted therapy, as both conditions present with contrast-enhancing lesions and perilesional edema on MRI. Advanced neuroimaging techniques, such as MR spectroscopy, perfusion MRI, and PET-CT, can improve diagnostic specificity by evaluating metabolic and vascular changes (46). Additionally, routine molecular profiling may help identify high-risk patients early, facilitating preemptive intervention.
The combination of clinical symptomatology and multimodal imaging enhances the accuracy of RN diagnosis, enabling differentiation from tumor progression and guiding therapeutic decisions. However, challenges remain in correlating imaging findings with clinical symptoms due to the complex pathophysiology and heterogeneous presentation of RN. Rodent models have provided insights into cellular and molecular mechanisms but do not fully replicate human clinical and radiological features, underscoring the need for further translational research to optimize diagnostic criteria and imaging biomarkers for RN. Given these complexities, multidisciplinary collaboration involving oncologists, radiation oncologists, and neurologists is essential for optimizing diagnosis and management.
Strategies to mitigate combined toxicity and enhance vascular repair
8
Treatment timing and sequencing
8.1
A strategic treatment hiatus is the most widely recommended mitigation strategy. Holding the BRAFi for 3–5 days before and after each RT fraction, particularly SRS, allows a window for the initiation of endogenous vascular repair mechanisms without the inhibitory pressure of the drug, significantly reducing the incidence of severe necrosis (47).
Rational drug regimen choice
8.2
Combining a BRAFi with a MEK inhibitor (MEKi) is a standard-of-care approach that not only improves antitumor efficacy but also reduces the skin and normal tissue toxicity associated with paradoxical MAPK activation. The MEKi blunts the ERK activation downstream of paradoxical RAF dimerization, thereby mitigating this key driver of pathologic signaling in normal tissues (48).
Optimized radiotherapy design
8.3
Employing hypofractionated RT schedules instead of single-fraction SRS can distribute the vascular injury over time, allowing for repair between fractions. Furthermore, employing highly conformal techniques and adhering to strict normal tissue dose constraints (e.g., for brainstem or hippocampi) can minimize the volume of sensitive tissue exposed to high doses.
Active management of established necrosis
8.4
Clinicians must be prepared to manage necrosis aggressively. High-dose corticosteroids are first-line to reduce edema. For steroid-refractory cases, bevacizumab (a VEGF-neutralizing antibody) has shown efficacy in numerous studies. By neutralizing the pathologic, leaky-vessel-inducing VEGF, it can reduce edema and improve radiographic appearance, though its use remains off-label (49).
Vigilant monitoring and adaptive treatment
8.5
Early detection is key. Frequent surveillance MRI with advanced sequences (e.g., perfusion-weighted imaging) is crucial to distinguish necrosis from progression. A low threshold for neurologic assessment and prompt adjustment of systemic therapy upon suspicion of necrosis can prevent irreversible neurological deficits.
Investigational pro-angiogenic and pro-repair strategies
8.6
Paradoxically, after the acute anti-cancer treatment phase, strategies to promote healthy angiogenesis may be beneficial for repairing RT-induced damage. The secretome of mesenchymal stem cells (MSCs) is rich in pro-angiogenic, anti-inflammatory, and trophic factors that can promote the formation of stable vasculature and mitigate inflammation. MSC-derived exosomes are under investigation as a cell-free therapy to stimulate vascular regeneration and repair long-term tissue damage after oncologic therapy (50).
Conclusion and future perspectives
9
In conclusion, the combination of BRAFi and RT for BRAF-mutant lung cancer brain metastases is a potent but double-edged sword, offering improved tumor control at the cost of increased RN risk. This review underscores the need for strategic BRAFi -RT integration, personalized dose adaptation, and vigilant monitoring. This interplay between RN and BRAFi effects necessitates a comprehensive understanding of molecular and cellular dynamics to inform clinical decision-making. Clinically, optimizing treatment regimens requires meticulous integration of molecular tumor profiling with advanced radiotherapeutic techniques. Tailoring the timing, dosage, and sequencing of targeted agents alongside precision radiotherapy modalities such as stereotactic radiosurgery or intensity-modulated radiotherapy can mitigate the risk of RN. Moreover, early pharmacologic interventions aimed at neuroprotection should be explored and incorporated into therapeutic protocols to safeguard neural tissue without compromising oncologic efficacy.
Looking forward, future research must prioritize elucidating the intricate molecular mechanisms underpinning RN, particularly the interactions between targeted therapies and radiation at the cellular and microenvironmental levels. Additionally, clinical trials evaluating innovative combination strategies that balance tumor control with neurotoxicity prevention are imperative. This review highlights the critical need for continued translational research and clinical innovation to achieve this balance and improve the prognosis for this challenging patient cohort.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nana FA Ocak S . Targeting BRAF activation as acquired resistance mechanism to EGFR tyrosine kinase inhibitors in EGFR-mutant non-small-cell lung cancer. Pharmaceutics. (2021) 13(9):1478. doi: 10.3390/pharmaceutics 13091478, PMID: 34575554 PMC 8471192 · doi ↗ · pubmed ↗
- 2Liu D Ding K Yin K Peng Z Li X Pan Y . A real world analysis of secondary BRAF variations after targeted therapy resistance in driver gene positive NSCLC. Sci Rep. (2024) 14:20302. doi: 10.1038/s 41598-024-71143-6, PMID: 39218919 PMC 11366755 · doi ↗ · pubmed ↗
- 3Sayin SI Eklund EA Ali KX Dzanan JJ Xylander M Dankis M . Distinct metastatic organotropism shapes prognosis in lung adenocarcinoma with brain metastasis. Front Oncol. (2025) 15:1569517. doi: 10.3389/fonc.2025.1569517, PMID: 40291914 PMC 12031661 · doi ↗ · pubmed ↗
- 4Souza VGP Telkar N Lam WL Reis PP . Comprehensive analysis of lung adenocarcinoma and brain metastasis through integrated single-cell transcriptomics. Int J Mol Sci. (2024) 25:24. doi: 10.3390/ijms 25073779, PMID: 38612588 PMC 11012108 · doi ↗ · pubmed ↗
- 5Mehta GU Raza SM . Management of skull base metastases. Neurosurg Clin N Am. (2020) 31:659–66. doi: 10.1016/j.nec.2020.06.013, PMID: 32921360 · doi ↗ · pubmed ↗
- 6Gondi V Bauman G Bradfield L Burri SH Cabrera AR Cunningham DA . Radiation therapy for brain metastases: an ASTRO clinical practice guideline. Pract Radiat Oncol. (2022) 12:265–82. doi: 10.1016/j.prro.2022.02.003, PMID: 35534352 · doi ↗ · pubmed ↗
- 7El Shafie RA Böhm K Weber D Lang K Schlaich F Adeberg S . Palliative radiotherapy for leptomeningeal carcinomatosis-analysis of outcome, prognostic factors, and symptom response. Front Oncol. (2019) 8:641. doi: 10.3389/fonc.2018.00641, PMID: 30671384 PMC 6331444 · doi ↗ · pubmed ↗
- 8Deng G Zhang Q Fan J Zhao C Jiao H Li Z . Optimal intervention timing for craniocerebral radiotherapy in EGFR mutant lung adenocarcinoma patients with brain metastases. BMC Cancer. (2024) 24:1571. doi: 10.1186/s 12885-024-13363-7, PMID: 39716108 PMC 11664826 · doi ↗ · pubmed ↗