Digenic sarcomeric variants in paediatric dilated cardiomyopathy and maternal peripartum cardiomyopathy: a familial case report
Hakan Kurt, Zülal Ülger Tutar, Ertürk Levent, Burcugül Karasulu Beci, Eser Doğan

TL;DR
A child with heart disease and her mother with postpartum heart issues share genetic variants linked to heart conditions, suggesting a family pattern.
Contribution
This case report identifies digenic sarcomeric variants in a family with pediatric and maternal cardiomyopathy.
Findings
A child and her mother share a MYH7 variant linked to different heart conditions.
The family exhibits phenotypic variability with distinct cardiomyopathy presentations.
Digenic variants may contribute to inherited cardiomyopathy syndromes.
Abstract
Left ventricular non-compaction (LVNC) can be observed as a phenotypic trait in patients with dilated cardiomyopathy. Familial cases have been increasingly recognized, with sarcomeric gene mutations—particularly in MYH7 and MYBPC3—playing a significant role. Peripartum cardiomyopathy (PPCM) may also share overlapping genetic architecture with inherited cardiomyopathies. We report a 7-year-old girl with a clinical diagnosis of dilated cardiomyopathy with LVNC since infancy. Genetic analysis revealed two heterozygous missense variants in sarcomeric genes associated with inherited cardiomyopathies: MYH7: c.4186C>T (p.Arg1396Trp) and MYBPC3: c.2672G>A (p.Arg891Gln), both classified as variants of uncertain significance. Segregation analysis showed that the MYH7 variant was maternally inherited and the MYBPC3 variant paternally inherited. Notably, the mother developed PPCM 4 months…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
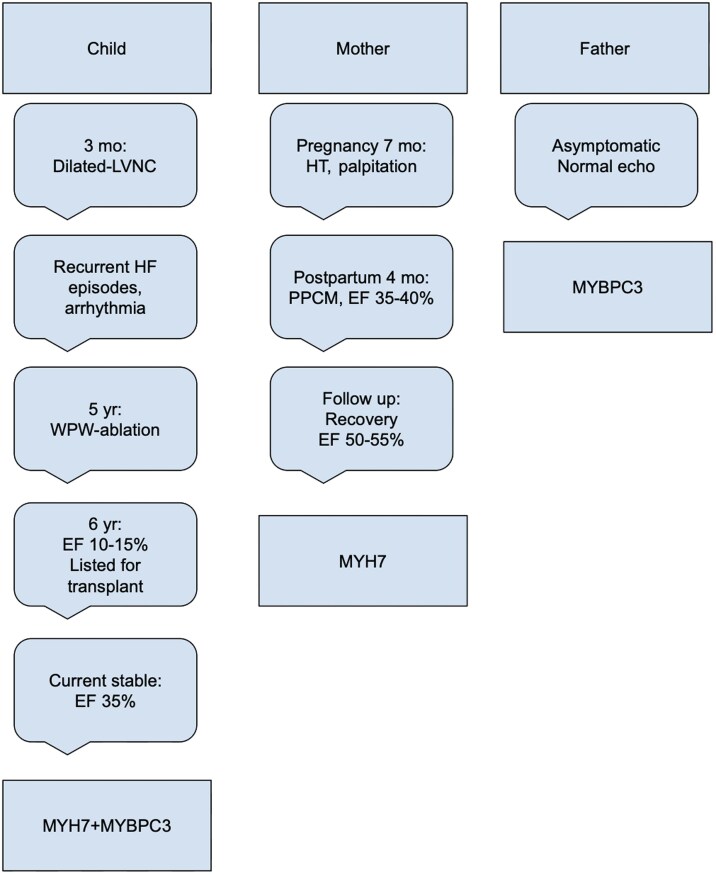 Figure 1
Figure 1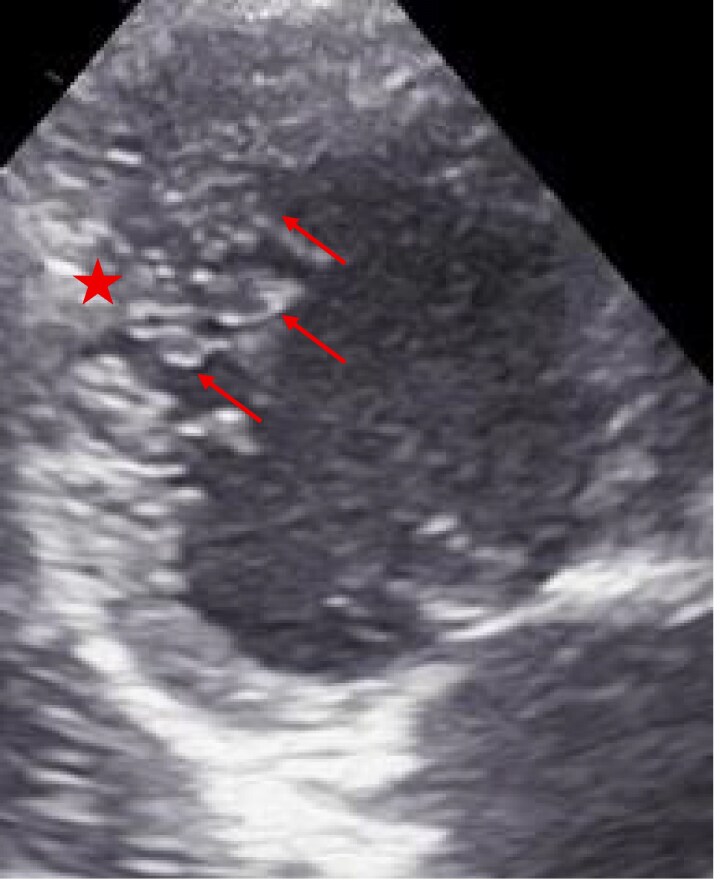 Figure 1
Figure 1 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Nuclear Structure and Function · Congenital heart defects research
Introduction
Left ventricular non-compaction (LVNC) is characterized by a prominent trabecular meshwork, a thin compacted layer, and deep intertrabecular recesses.^1^ Left ventricular non-compaction shows incomplete penetrance and variable expressivity, spanning prenatal/neonatal onset to asymptomatic adulthood, and is familial in ∼30% of cases.^2^ Genetically, defects disrupting myocardial compaction—most often in sarcomeric or related genes (ACTC1, MYH7, MYBPC3, TNNT2, TPM1, TTN, LDB3, LMNA, DTNA)—are frequently implicated.^2,3^ Diagnosis relies on imaging; Jenni’s echocardiographic criteria use an end-systolic non compacted/compacted ratio (NC/C ratio) >2, while magnetic resonance imaging (MRI)/computed tomography (CT) studies suggest a diastolic NC/C ratio >2.3 to improve specificity and distinguish pathology from physiologic hypertrabeculation.^4^ Peripartum cardiomyopathy presents with reduced LVEF late in pregnancy or early postpartum and can overlap genetically with familial dilated cardiomyopathy (DCM)/LVNC; familial clustering and DCM-gene variants support selective genetic testing.^5^ Up to 20% of PPCM patients harbour pathogenic variants—most commonly TTN truncations—while sarcomeric genes such as MYH7 and MYBPC3 also contribute via impaired structure/contractility.^6,7^
Summary figure
**
Case presentation
A 7-year-old girl, diagnosed with DCM with LVNC at 3 months, was born at term by caesarean section with normal perinatal course and age-appropriate neurodevelopment. She experienced recurrent heart failure and supraventricular tachycardia with intermittent Wolff–Parkinson–White; at age 5, electrophysiology confirmed a right posterolateral accessory pathway and successful ablation. At 6 years, transplant evaluation showed prominent LV trabeculation with NC/C ratio 2.6 and LVEF 10%–15% (Figures 1 and 2). Work-up was negative for metabolic/infectious aetiologies; CT angiography showed cardiomegaly, biventricular dilation, and increased trabeculation. She was listed for transplantation and closely followed. Targeted next-generation sequencing identified heterozygous missense variants in MYH7 (NM_000257.4:c.4186C>T, p.Arg1396Trp) and MYBPC3 (NM_000256.3:c.2672G>A, p.Arg891Gln); segregation showed maternal inheritance of MYH7 and paternal inheritance of MYBPC3. The non-consanguineous mother (30 years) developed gestational hypertension and tachycardia; 4 months postpartum she presented with dyspnoea/palpitations and was diagnosed with PPCM (LVEF 35%–40%); on follow-up, her LVEF improved to 50%–55% with mild global hypokinesia and increased trabeculation (NC/C ratio 1.4). The father (31 years) is asymptomatic with normal echocardiography. Genetic analysis was limited to the parents, as other family members were not accessible or available for testing. The child remains clinically stable on optimized therapy, with LVEF 30%–35%, and is listed for heart transplantation.
Apical long-axis view from transthoracic echocardiography demonstrating prominent trabeculations and deep intertrabecular recesses within the left ventricular apex, consistent with morphological features of left ventricular non-compaction. Red arrows highlight the regions of prominent trabeculation. The red asterisk indicates the compact myocardial layer.
Short-axis echocardiographic view showing excessive trabeculations and deep intertrabecular recesses in the left ventricular myocardium, suggestive of left ventricular non-compaction. Red arrows highlight the regions of prominent trabeculation. The red asterisk indicates the compact myocardial layer.
Discussion
Recent genetic studies highlight the role of sarcomeric mutations, particularly in MYH7 and MYBPC3, in LVNC, DCM, and PPCM. Pathogenic variants in these genes are found in about 25% and 13% of LVNC cases, respectively, with a domain-specific correlation: MYH7 head-domain variants associate with isolated LVNC and fewer adverse events, while tail-domain variants more often link to an LVNC/DCM overlap and may disrupt titin binding, mimicking TTN effects.^8^ In a large MYH7-related DCM cohort, LVNC features were common across ages, reinforcing the LVNC-DCM overlap.^9^ Although often milder in adult-onset LVNC, MYH7 mutations can cause severe perinatal disease, as shown in foetuses with hydrops fetalis from maternal mosaicism and in infants with biventricular non-compaction and restrictive physiology, both leading to early heart failure and death.^2,10^ Overall, pathogenic sarcomeric variants—especially compound heterozygous or digenic states—occur in up to 22% of LVNC and are linked to more severe phenotypes, suggesting additive or modifying effects.^3,11^
In another report, digenic inheritance involving MYH7 and LAMA4 was linked to severe early-onset DCM, with the LAMA4 variant acting as a genetic modifier.^12^ Similarly, Petropoulou et al.^13^ described a consanguineous family with dual MYH7 and TNNT2 variants, where only individuals carrying both mutations exhibited disease, reinforcing the concept of synergistic genetic interactions.
Our paediatric case presents with a severe infant-onset DCM with LVNC phenotype in the setting of dual heterozygous missense variants in MYH7 (c.4186C>T; p.Arg1396Trp) and MYBPC3 (c.2672G>A; p.Arg891Gln), inherited maternally and paternally, respectively. The mother, who carries only the MYH7 variant, was diagnosed with PPCM 4 months postpartum, displaying a milder but clinically significant phenotype with reduced ejection fraction. In contrast, the father—carrier of the MYBPC3 variant—remains asymptomatic with normal cardiac imaging.
The MYBPC3 c.2672G>A (p.Arg891Gln) variant is a missense change classified in ClinVar as a Variant of Uncertain Significance (VUS) due to limited evidence and is not listed in OMIM as directly linked to LVNC. Although its pathogenicity is uncertain, MYBPC3 is a well-established cardiomyopathy gene, particularly in hypertrophic cardiomyopathy (HCM), with some variants also reported in LVNC.
The MYH7 c.4186C>T (p.Arg1396Trp) variant, likewise listed in ClinVar as a VUS and not individually associated with LVNC or DCM in OMIM, affects the β-myosin heavy chain. While this specific change lacks direct evidence, MYH7 is extensively implicated in HCM, DCM, and LVNC.
In our patient, the MYH7 variant is non-truncating and localized to the tail domain, consistent with the LVNC/DCM overlap phenotype. As reported by van Waning et al.,^8^ such tail-domain variants may disrupt titin binding and phenocopy the effects of TTNtv, conferring a higher risk of systolic dysfunction. These features further support the need for a genotype-guided approach to management and surveillance in cardiomyopathy patients, particularly in the peripartum context.
The early-onset and more severe clinical presentation in our case may reflect a potential synergistic or modifying effect of carrying both variants, supporting a hypothesis of cumulative pathogenicity or digenic interaction. In addition, this familial observation illustrates the need for long-term cardiologic follow-up of the asymptomatic father, given his carrier status of a sarcomeric gene variant (MYBPC3) that may yet lead to a late-onset phenotype.
In PPCM, growing evidence supports its overlap with familial DCM, particularly in patients harbouring pathogenic variants in cardiomyopathy-associated genes. Multiple studies have shown that 15%–20% of PPCM patients carry variants in genes such as TTN, MYH7, MYBPC3, LMNA, and SCN5A. Pregnancy-related stressors may unmask subclinical cardiomyopathy in gene-positive, asymptomatic women, leading to overt heart failure.^14,15^
van Spaendonck-Zwarts et al.^5^ conducted a foundational study in families with coexisting PPCM and DCM, revealing pathogenic mutations in over half of the families. Familial PPCM patients showed significantly lower recovery rates of left ventricular function compared to general PPCM populations, suggesting genetic predisposition may confer worse outcomes.
Our case further supports the notion that PPCM may be a manifestation of underlying genetic predisposition, potentially triggered or unmasked by the haemodynamic stresses of pregnancy. The presence of a shared variant between mother and child, manifesting as distinct cardiomyopathy phenotypes, underscores the variable expressivity often seen in sarcomeric gene-related cardiomyopathies.
Conclusion
This case highlights the complexity of genotype–phenotype relationships in inherited cardiomyopathies and underscores the clinical relevance of digenic sarcomeric variants in early-onset disease. The co-occurrence of paediatric DCM with LVNC phenotype and maternal PPCM within the same family supports a possible role for MYH7 variants in both phenotypes and raises the need to consider inherited cardiomyopathy even in apparently sporadic cases. These findings emphasize the importance of comprehensive family screening, careful variant interpretation in clinical context, and long-term surveillance of asymptomatic carriers. Moreover, further functional studies are warranted to clarify the pathogenic potential of variants that currently fall below established classification thresholds but may still contribute meaningfully to disease expression.
Lead author biography
2010–16: Medical Education, Istanbul University, Cerrahpaşa, Faculty of Medicine. 2018–23: Residency in Paediatrics, Mersin University, Department of Pediatrics. June 2024–present: Fellowship in Paediatric Cardiology, Ege University, Division of Pediatric Cardiology
Consent: Written informed consent was obtained from the patient’s legal guardians for the publication of this case report and any accompanying images.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jaouadi H, El Louali F, Wanert C, Cano A, Ovaert C, Zaffran S. Dilated-left ventricular non-compaction cardiomyopathy in a pediatric case with SPEG compound heterozygous variants. Int J Mol Sci 2022;23:5205.35563595 10.3390/ijms 23095205 PMC 9102709 · doi ↗ · pubmed ↗
- 2Kawamura H, Ikawa M, Hirono K, Kimura J, Okuno T, Kawatani M, et al Low-frequency maternal novel MYH 7 mosaicism mutation in recurrent fetal-onset severe left ventricular non-compaction: a case report. Front Pediatr 2023;11:1195222.37360367 10.3389/fped.2023.1195222 PMC 10285293 · doi ↗ · pubmed ↗
- 3Sedaghat-Hamedani F, Haas J, Zhu F, Geier C, Kayvanpour E, Liss M, et al Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur Heart J 2017;38:3449–3460.29029073 10.1093/eurheartj/ehx 545 · doi ↗ · pubmed ↗
- 4Yu W, Thomas MA, Mills L, Wright JR Jr. Prenatal diagnosis of isolated right ventricular non-compaction cardiomyopathy with an MYH 7 likely pathogenic variant. Fetal Pediatr Pathol 2023;42:464–471.36630130 10.1080/15513815.2022.2120785 · doi ↗ · pubmed ↗
- 5van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, et al Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J 2014;35:2165–2173.24558114 10.1093/eurheartj/ehu 050 · doi ↗ · pubmed ↗
- 6Sliwa K, Bauersachs J, Arany Z, Spracklen TF, Hilfiker-Kleiner D. Peripartum cardiomyopathy: from genetics to management. Eur Heart J 2021;42:3094–3102.34322694 10.1093/eurheartj/ehab 458 · doi ↗ · pubmed ↗
- 7Spracklen TF, Chakafana G, Schwartz PJ, Kotta MC, Shaboodien G, Ntusi NAB, et al Genetics of peripartum cardiomyopathy: current knowledge, future directions and clinical implications. Genes (Basel) 2021;12:103.33467574 10.3390/genes 12010103 PMC 7830587 · doi ↗ · pubmed ↗
- 8van Waning JI, Moesker J, Heijsman D, Boersma E, Majoor-Krakauer D. Systematic review of genotype–phenotype correlations in non-compaction cardiomyopathy. J Am Heart Assoc 2019;8:e 012993.31771441 10.1161/JAHA.119.012993 PMC 6912966 · doi ↗ · pubmed ↗