Chemical-mediated alteration of antibiotic susceptibility: mechanisms and potential new targets for antibiotic adjuvant discovery
Joshua M. E. Adams, Omar M. El-Halfawy

TL;DR
This paper reviews how small molecules at infection sites can influence antibiotic effectiveness and explores new targets for improving antibiotic treatments.
Contribution
The paper highlights chemical-mediated resistance mechanisms and identifies potential new targets for antibiotic adjuvant development.
Findings
Small molecules at infection sites can transiently alter bacterial responses to antibiotics.
Chemicals produced by bacteria may offer communal protection against antibiotics.
Genetically encoded mechanisms for chemical response could serve as new drug targets.
Abstract
The critical rise in antimicrobial resistance rates, coupled with a nearly stagnant drug discovery pipeline, has resulted in a sharp surge in clinical failure of antibiotic therapy. Antimicrobial resistance, possibly leading to therapeutic failures, has been predominantly attributed to genetically encoded intrinsic or acquired resistance determinants. However, small molecules that bacteria may be exposed to at the infection site have been implicated in antibiotic resistance, tolerance, and persister formation. Such chemicals, either produced by the host or bacteria, typically elicit transient effects on antibiotic responses while bacteria are exposed to them; as such, their effects may alter therapeutic outcomes but would not be detected in standard in vitro antibiotic susceptibility tests. Chemicals produced by certain bacteria may alter the response of the same or other bacterial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
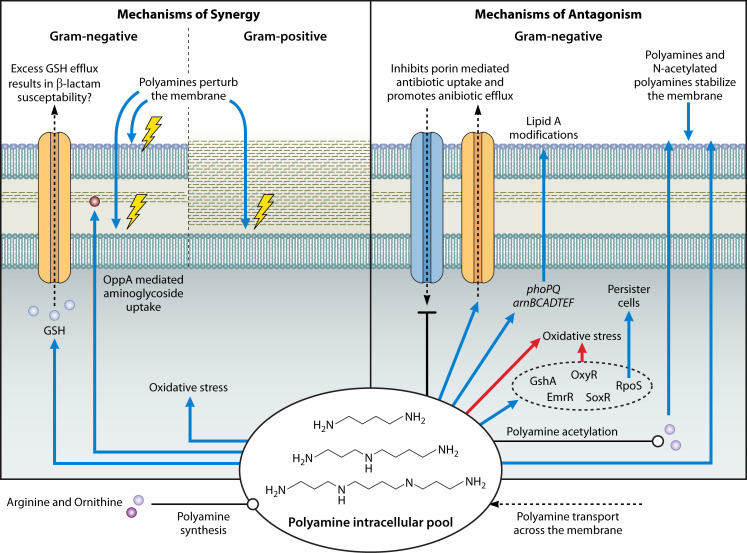 Fig 1
Fig 1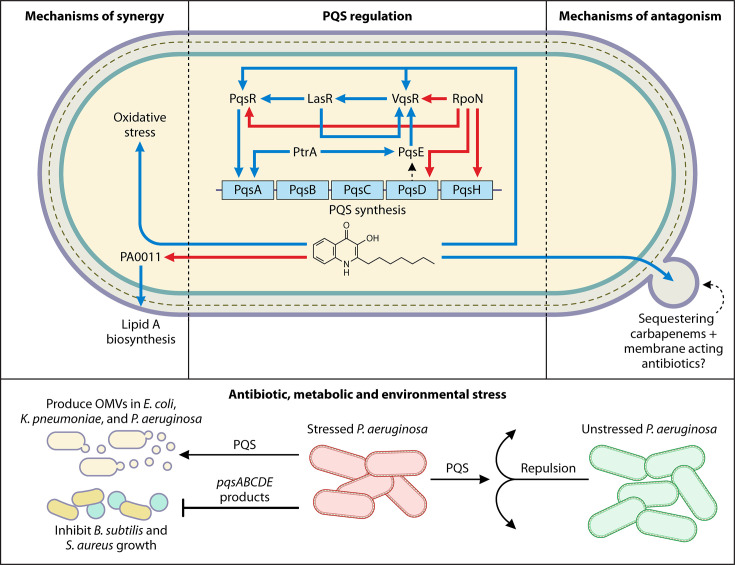 Fig 2
Fig 2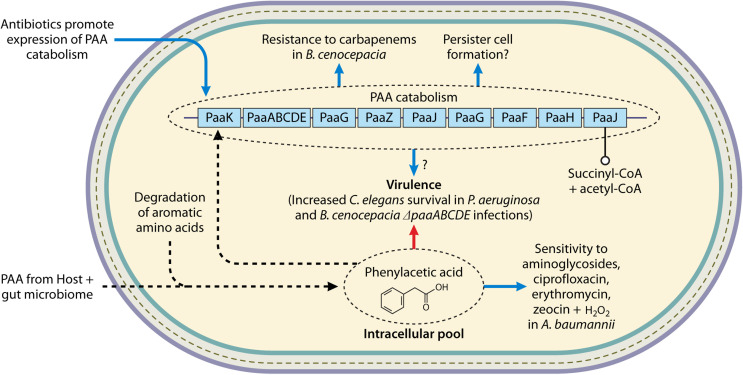 Fig 3
Fig 3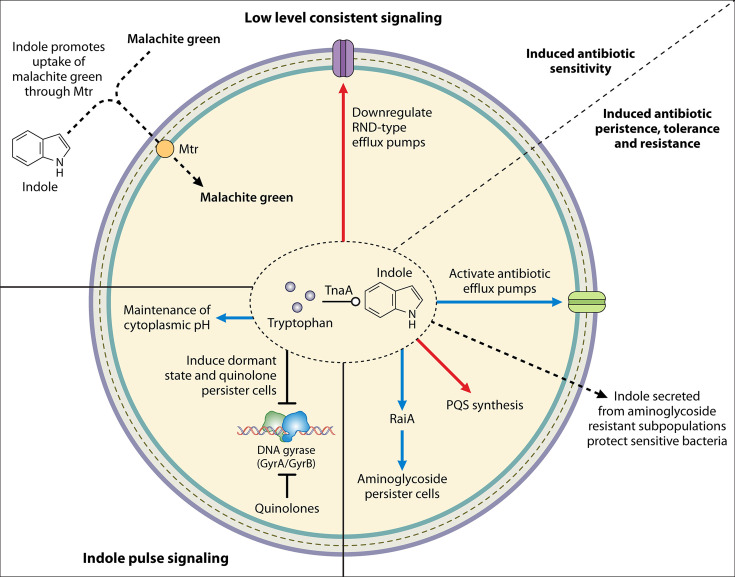 Fig 4
Fig 4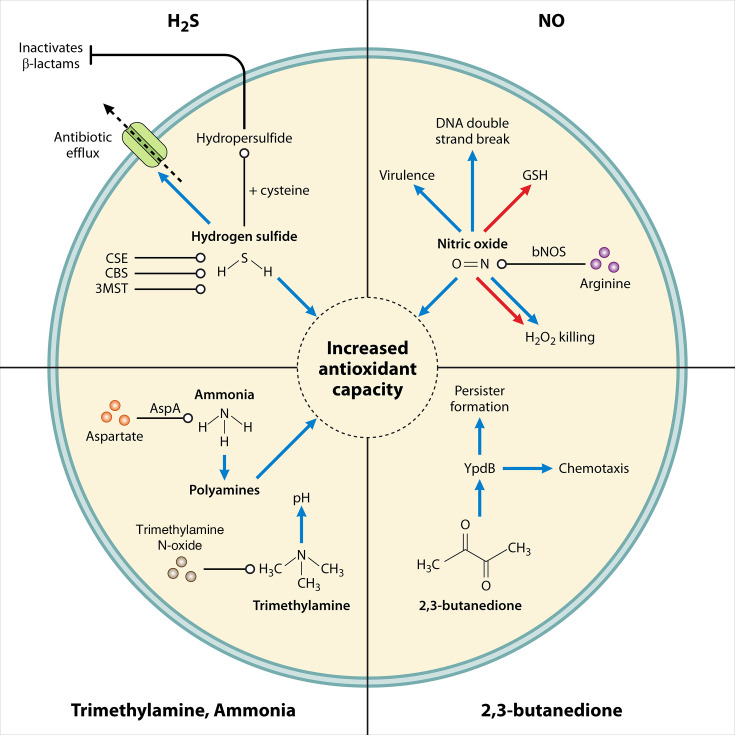 Fig 5
Fig 5| Target | Identified inhibitors, analogs, and delivery vectors | Potentiated antibiotics or | References |
|---|---|---|---|
| Polyamines | |||
| Ornithine decarboxylase | Difluoromethylornithine | Polymyxin B (PMB) | ( |
| SpeG | Diminazene | Tetracycline, ciprofloxacin, vancomycin, kanamycin, and rifampicin. | ( |
| Polyamine uptake in | 44-Ant-44 | Prolongs survival of | ( |
| Membrane and ribosome | Polyamine analogs N1-Dodecylpropane-1,3-diamine (OES2-017) Octan-1-amine (OES2-046) (3-aminopropyl)-(octyl)amine (OES2-045) Dioctylamine (OES2-008) | Azithromycin. | ( |
| PQS | |||
| PqsR | Quinazoline disulfide analog | Tobramycin, PMB, levofloxacin, kanamycin, and ciprofloxacin | ( |
| PqsR/PqsBC | Nonligand-based benzamide–benzimidazole analog | Meropenem | ( |
| PqsR/PqsD | Compound 6 | Ciprofloxacin | ( |
| PqsA | Norharmane | Carbapenems and PMB | ( |
| PQS | Improved survival of a mouse model co-infected with | ( | |
| PAA | |||
| PaaE | Deletions/disruptions in | ( | |
| PaaB | Deletions in | ( | |
| PaaA | Insertion disruptions in | ( | |
| Indole | |||
| TnaA | ALG-05 | Deletions in | ( |
| Central carbon and nitrogen metabolism-linked metabolites | |||
| Proton motive force | Pyruvate, inosine, fructose, glucose, mannitol, alanine, citrulline, and glutamine. | Aminoglycosides, tetracyclines, and β-lactam. | ( |
| Porins OmpF, OmpK 36 | Glutamine | β-lactams and tetracycline. | ( |
| Reactive oxygen species generation | Pyruvate | Aminoglycosides and cefoperazone-sulbactam. | ( |
| VOC | |||
| 3-mercaptopyruvate sulfurtransferase | Aspartate | Ampicillin. | ( |
| SseA | Deletions lead to lower | ( | |
| Cystathionine β-synthase, cystathionine γ-lyase | DL-propargylglycine | Gentamicin, ampicillin, norfloxacin, and amikacin. | ( |
| Cystathionine γ-lyase | NL1 | Gentamicin in | ( |
| H2S sequestering delivery vector Gm@UiO-66-MA | Gentamicin. | ( | |
| H2S scavenger 7B | Gentamicin. | ( | |
| NO-containing implant devices | Reduce bacterial cell viability and disperse biofilms. | ( | |
| PurA | Aurodox | Aurodox has non-sufficient target selectivity. Δ | ( |
- —Canadian Institutes of Health Researchhttp://dx.doi.org/10.13039/501100000024
- —Canadian Institutes of Health Researchhttp://dx.doi.org/10.13039/501100000024
- —Natural Sciences and Engineering Research Council of Canadahttp://dx.doi.org/10.13039/501100000038
- —Natural Sciences and Engineering Research Council of Canadahttp://dx.doi.org/10.13039/501100000038
- —Cystic Fibrosis Canadahttp://dx.doi.org/10.13039/501100000082
- —Saskatchewan Health Research Foundationhttp://dx.doi.org/10.13039/501100000106
- —Canada Research Chairshttp://dx.doi.org/10.13039/501100001804
- —New Frontiers in Research Funds
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial agents and applications · Phenothiazines and Benzothiazines Synthesis and Activities · Antimicrobial Peptides and Activities
INTRODUCTION
The overuse and misuse of antibiotics in the interconnected One Health human, animal, and environmental triad have accelerated the emergence of antimicrobial resistance (AMR) (1). This critical rise in AMR, alongside the concurrent slow and expensive drug discovery pipeline, has resulted in a sharp rise in clinical failure rates of antibiotic therapy (2, 3). If the current trends continue, AMR has been projected to cause an attributable cumulative death toll of ~40 million deaths worldwide between the years 2025 and 2050 (4).
Antibiotic therapeutic failures are often ascribed to conventional resistance mechanisms, yet emerging evidence indicates that small molecules present at the infection site can influence antibiotic resistance, tolerance, and persister formation. Bona fide resistance determinants such as specific antibiotic-modifying enzymes, efflux pumps, and impermeable cell membranes lead to stable, high levels of antibiotic resistance (5, 6). Efforts to combat AMR have predominantly focused on defining and targeting such resistance mechanisms (6, 7). However, a dynamic network of signaling compounds and other small molecules—secreted by bacteria or produced by mammalian host cells—has been shown to influence bacterial responses to antibiotics (8). These chemical-mediated effects often lead to transient AMR occurring while bacteria are exposed to such small molecules, which may still alter the antibiotic therapeutic outcomes, potentially underlying some of the antibiotic therapeutic failures. Notably, the effects of many of the chemicals likely present at infection sites on bacterial responses to antibiotics are typically not captured under the conditions of standard antimicrobial susceptibility testing (AST) assays, which may explain, in part, some instances of discordance between AST results and antibiotic therapeutic outcomes (9).
Targeting biosynthesis, uptake, and response mechanisms to such chemicals expands the potential list of druggable targets; inhibitors of such targets may serve as antibiotic adjuvants that improve therapeutic outcomes of current antibiotic treatments. These potential targets are typically non-essential and are involved in antibiotic resistance as a result of the chemical landscape at the infection site. Given that inhibitors thereof would suppress bacterial growth only when combined with antibiotics, resistance to these drug combinations could still be derived from selective pressure at the site of infection. Resistance to various antibiotic adjuvants has been detected; for example, resistance to various β-lactam antibiotic adjuvants, including β-lactamase inhibitors (reviewed in reference 10) and an inhibitor of the envelope stress response regulator GraR (11), was more readily selected for upon exposure of bacteria to the antibiotic-antibiotic adjuvant combinations (10, 11). Yet, we posit that inhibitors acting as antibiotic adjuvants may have reduced selective pressure to drive resistance when present alone outside of the infection site in environmental settings. This feature may help mitigate the acquisition of resistance due to environmental exposure, which is a major contributor to the continual development of AMR (12).
This review will shed light on chemical-mediated alteration of antimicrobial susceptibility and the molecular basis thereof, covering the significant advancements made in this area since we reviewed it in 2012 (8). Chemicals covered include polyamines, Pseudomonas quinolone signal (PQS), phenylacetic acid (PAA), indole, and several microbial-secreted volatile organic compounds (VOCs). We also provide only select examples of the effects of central carbon and nitrogen metabolism-linked metabolites on antibiotic susceptibility, as this topic has recently been reviewed and examined from a metabolic-reprogramming perspective (13). Of note, bacterial cell-to-cell communication mediated by acyl homoserine lactone quorum-sensing signals has been previously extensively reviewed and will not be discussed in this article (14–18). Additionally, we will highlight the progress made in the identification of novel potential drug targets and the discovery of antibiotic adjuvant compounds as a result of the mechanistic elucidation of chemical-mediated alteration of antimicrobial responses.
CHEMICAL-MEDIATED ALTERATION OF ANTIBIOTIC SUSCEPTIBILITY
In this section, we will discuss different small molecules that elicit alteration of antibiotic susceptibility and the molecular basis thereof. We will attempt to identify novel targets that can be exploited for antimicrobial adjuvant therapy and progress made in identifying inhibitors of these targets (Table 1).
Polyamines
The biogenic polyamines spermidine, spermine, putrescine, and cadaverine are small polycationic molecules ubiquitous in nature. The polyamine structure consists of primary and secondary amine groups spaced by hydrophobic hydrocarbon bridges. This unique structure of polyamines creates favorable interactions with macromolecules in living organisms, where they influence RNA transcription, chromosomal structure, protein synthesis, acid resistance, biofilm formation, signal cellular differentiation, and can scavenge for free radical ions (71, 72). Natural polyamines are involved in the host immune responses, inhibiting macrophage activation, inducing autophagy, and suppressing necrosis (73, 74). Spermine, spermidine, and putrescine are produced at higher levels by regenerating tissues and released by dying cells into the extracellular milieu at the infection site (73, 75), resulting in an accumulation of polyamines, reaching concentrations within the millimolar range, dependent on the infection location (75–78). Such polyamine accumulation was observed in Pseudomonas aeruginosa infections of the sinuses, lungs, and in the sputum of cystic fibrosis patients, as well as in Staphylococcus aureus abscess infections (75, 78). Another manifestation of the potential influx of polyamines at the infection site can be inferred from an iterative comparative metabolomics study of common gram-negative pathogens found in bloodstream infections (Escherichia coli, Klebsiella spp., and Pseudomonas spp.) that revealed that polyamines acetylated by polyamine acetyltransferases are elevated during infection (22).
Polyamine alteration of antibiotic responses
Several studies reported polyamine-mediated protection from antibiotic stress. Bactericidal antibiotics were shown to elicit oxidative stress, which may contribute in part to their lethality (79). One mechanism of the polyamine involvement in bacterial antibiotic stress response is alleviating the oxidative stress resultant from bactericidal antibiotic exposure (19, 80, 81). Endogenous polyamine biosynthesis increases in response to antibiotic-induced oxidative stress in bacteria, including E. coli and Burkholderia cenocepacia, where the promoters of genes encoding amino acid decarboxylases, namely ornithine decarboxylase (converting ornithine into putrescine) and lysine decarboxylase (producing cadaverine), are induced (19, 80, 81). Moreover, Pseudomonas syringae increases extracellular putrescine concentrations in response to H_2_O_2_ stress, and mutants that exhibit increased putrescine excretion have increased survival in the presence of H_2_O_2_ (82). However, P. aeruginosa, upon H_2_O_2_ stress, has conversely been shown to downregulate genes encoding polyamine biosynthesis enzymes (83). Exogenous supplementation of either putrescine or spermidine reduces intracellular reactive oxygen species (ROS) in E. coli, B. cenocepacia, and P. syringae (19, 81, 82, 84–87). Moreover, depriving bacteria of endogenous and exogenous polyamines, which can lead to growth defects, reduced their ability to cope with oxidative stress; for example, various polyamine biosynthesis deletion mutants of E. coli and P. syringae were rapidly killed by ROS when grown in polyamine-deplete media such as polyamine-free Vogel-Bonner medium (82, 85, 86, 88). Perturbation of putrescine synthesis in B. cenocepacia and spermidine synthesis in P. aeruginosa leads to an increase in ROS generation upon polymyxin B (PMB) stress (19, 89); an increase in membrane lipid peroxidation was further observed in P. aeruginosa (89). Furthermore, the deletion of spermidine synthesis in Shigella spp. results in a larger H_2_O_2_ zone of inhibition when grown on minimal media (90).
Polyamines alleviate oxidative stress through direct and indirect mechanisms. Spermine and spermidine can quench singlet oxygen, and all polyamines can scavenge hydroxyl and superoxide radicals (19, 91–93). Additionally, putrescine exerts its antioxidant protection effects by increasing the transcription of rpoS in E. coli and oxyR in B. cenocepacia and E. coli, and the overexpression of SoxR, RpoS, EmrR, and GshA in E. coli (86, 87, 94–97). RpoS is a general stress response regulator that responds to peroxides via induction of catalases KatE and KatG; genes encoding both were upregulated upon putrescine exposure (87). Additionally, micromolar concentrations of exogenous spermidine and putrescine induced the rpoS promoter and increased RpoS expression, which was shown to be responsible for an increase in persister cell formation in netilmicin-treated E. coli (95, 97). OxyR is a direct sensor of hydrogen peroxide stress that induces the expression of catalases and peroxidases, whereas SoxRS detoxifies superoxides via regulation of a >100 gene regulon that includes the superoxide dismutase SodA (98). Upregulation of GshA directly increases glutathione (GSH) synthesis, while EmrR, a negative transcriptional factor for multidrug efflux pumps, was hypothesized to indirectly increase GSH synthesis via inhibition of the efflux of GSH precursors (87). The resulting increase in GSH peroxidases detoxifies hydrogen peroxides (87). In contrast, excessive polyamines can promote toxic accumulation of superoxide radicals in E. coli and create improper cellular redox states in Acinetobacter baumannii (99–102). Polyamine-induced efflux of GSH was hypothesized to be a contributing factor to polyamine-β-lactam in vitro synergy against A. baumannii in checkerboard assays (101, 102). Together, polyamine homeostasis is fine-tuned in bacteria; the loss of biosynthesis or excess of polyamines can result in detrimental effects to the bacteria’s ability to manage oxidative stress and antibiotic insult.
Another mechanism of polyamine alteration of antibiotic susceptibility can be attributed to bacterial membrane effects. Membrane-level effects included changes to drug uptake and efflux, leading to polyamine-mediated protection from antibiotics. Polyamines have been shown to inhibit porin-mediated uptake of fluoroquinolone in Mycobacteria and E. coli and β-lactams in E. coli and P. aeruginosa (103–106). The modulation of porins was most pronounced by spermidine and spermine relative to other polyamines (103, 105, 106). Moreover, putrescine, spermidine, and cadaverine disrupt the interaction between the repressor AcrR and the acrAB promoter, resulting in induction of the multidrug efflux pump AcrAB-TolC (107). In Burkholderia pseudomallei, exogenous spermidine stimulated the bpeA promoter in promoter-reporter assays; BpeA is a component of the BpeAB-OprB efflux pump (108). In contrast, polyamine-mediated sensitization to antibiotics was observed, whereby increased levels of intracellular polyamines in E. coli led to aminoglycoside susceptibility by increasing the expression of OppA, a periplasmic protein involved in aminoglycoside uptake (109, 110).
Polyamine membrane effects also include their ability to perturb outer and inner membranes. Putrescine synergizes with macrolides in checkerboard assays against Klebsiella pneumoniae and E. coli via perturbation of the outer and inner membranes (25). Additionally, spermidine and spermine have been shown to displace Ca^2+^ from the outer membrane of E. coli to a similar extent as PMB nonapeptide (111). CRISPRi suppression of speG, encoding a polyamine acetyltransferase that detoxifies polyamines, in E. coli clinical isolates resulted in increased endogenous putrescine concentrations and decreased acetylated putrescine concentrations (22). These isolates exhibited increased outer and inner membrane permeability and increased intracellular antibiotic accumulation (22). However, it is unclear if this is a result of the heightened putrescine concentration, loss of acetylated putrescine, or other unrelated mechanisms (22).
Furthermore, polyamines can stabilize the outer membrane, increasing resistance to cationic peptides by up to 16-fold shift in the minimum inhibitory concentration (MIC) (19, 78, 112, 113). P. aeruginosa treated with spermidine exhibit induction of the two-component PhoPQ system and the arnBCADTEF operon, whose products mediate the addition of aminoarabinose to lipid A, a known mechanism for cationic peptide resistance in multiple gram-negative bacteria (113). Interestingly, exogenous polyamines also reduced cationic peptide-induced expression of arnC, suggesting that polyamines can compete with antimicrobial peptides for outer membrane binding (89). A membrane molecular dynamics simulation provided evidence that polyamines can stabilize the outer membrane via lipid A non-covalent crosslinks (114). Putrescine outcompeted PMB for surface binding on B. cenocepacia, contributing to putrescine-mediated protection from the antibiotic in wild-type and PMB-sensitive subpopulations of B. cenocepacia (19, 112). Polyamines at the infection site in vivo may also confer surface protection from cationic antimicrobial peptides; spermidine bound to the surface of P. aeruginosa wild type and ΔpmrB, shielding the membrane net negative charge and reducing the surface binding of PMB (78). P. aeruginosa lacking PmrB have impaired aminoarabinose modification of LPS, exhibiting an increased outer membrane negative charge and thus are more susceptible to PMB in vitro; however, in an in vivo sinus and lung murine infection model, these mutants do not exhibit bacterial load change when compared to the wild-type strain (78). The mechanisms of polyamine-mediated alteration to antibiotic susceptibility are summarized in Fig. 1.
Mechanisms of polyamine-mediated alteration of antibiotic susceptibility. Polyamines synergize with antibiotics by increasing membrane permeability in both gram-negative and gram-positive bacteria. Synergy is also mediated by polyamine promotion of superoxide accumulation and creation of improper cellular redox states via polyamine-induced GSH efflux. Conversely, polyamines antagonize antibiotics in gram-negative bacteria via inhibition of porin-mediated antibiotic uptake, promoting antibiotic efflux pumps, inducing expression of lipid A modification pathways to combat polymyxins, alleviating oxidative stress, and promoting persister cell formation. Additionally, polyamines, and potentially acetylated polyamines, stabilize the outer membrane to reduce antibiotic permeability. The polyamine intracellular pool in bacteria consists of endogenously produced and exogenously acquired polyamines. For detailed descriptions of each mechanism and the corresponding references, refer to the text. Blue arrows denote increased, red arrows denote decreased, blunt-head arrows denote inhibited, dotted arrows denote the movement of a substrate, and open circled arrows denote synthesis.
Potential targets based on the molecular mechanism of polyamines
Interfering with polyamine homeostasis and environmental polyamine availability may provide new potential antimicrobial strategies. One potential strategy is the inhibition of polyamine synthesis. The inhibition of spermidine synthesis in B. pseudomallei via dicyclohexylamine, a bacterial and eukaryotic polyamine synthesis inhibitor (115, 116), exhibited a reduction in erythromycin efflux proposed to be due to downregulation of the efflux pumps BpeAB-OprB, AmrAB-OprB, and BpeEF-OprC (108). Dicyclohexylamine also inhibited the growth of B. cenocepacia by 75% at half-MIC of PMB, likely by reducing polyamine protection from PMB (112). Likewise, targeting polyamine synthesis using the ornithine decarboxylase inhibitor difluoromethylornithine is effective against the parasite Trypanosoma brucei subspecies and the fungus Pneumocystis pneumonia (PCP) (20, 21). Additionally, targeting the availability of polyamines in infection environments may serve as a novel strategy to prevent polyamine-induced protection from current therapies. Inhibitors, such as N-(4-amino-butyl)-N′-(10-{[4-(4-amino-butylamino)butylamino]-methyl}nthracene-9-ylmethyl)butane-1,4-diamine (44-Ant-44) and N-(4-amino-butyl)-N′-(4-{[4-(4-amino-butylamino)butylamino]-methyl}benzyl)butane-1,4-diamine (44-Bn-44), reduced polyamine uptake in alveolar macrophages, reducing environmental polyamine availability and prolonging the survival of PCP-infected rats (24).
Targeting polyamine detoxification mechanisms, such as the spermine/spermidine acetyltransferase SpeG, may serve as another target for antibiotic adjuvant therapies. CRISPRi suppression of speG in E. coli increases sensitivity by eightfold to vancomycin, an antibiotic typically only utilized against gram-positive bacteria due to its poor penetration of the outer membrane (22). Diminazene, a structural mimic of spermidine used as an antitrypanosomal and antibabesial agent and also shown to inhibit the human polyamine N-acetyltransferase SAT1 and bacterial SpeG in E. coli (22, 117), increased the sensitivity of E. coli to vancomycin eightfold (22). Diminazene also re-sensitized multidrug-resistant E. coli, K. pneumoniae, and P. aeruginosa to tetracycline and ciprofloxacin in checkerboard assays in vitro and synergized with tetracycline in vivo in an E. coli cecal-slurry model of sepsis by increasing intracellular accumulation of the antibiotics (22). Recently, a polyamine analog has been identified as a novel broad-spectrum inhibitor of SpeG in S. aureus (SpeG^+^ strains) and other various gram-negative and gram-positive pathogens (23). This inhibitor, acting at low micromolar ranges, synergized with polyamines, lowering their bacterial growth inhibitory concentrations by 32-fold, abolished polyamine-mediated resistance to the antibiotics vancomycin, kanamycin, and rifampicin in vitro, and eradicated SpeG-expressing Salmonella enterica serovar Typhimurium inside murine macrophages (23). However, the utility of SpeG as an adjuvant target may be species and strain specific. Despite generally being non-essential for bacterial survival, SpeG was essential in two E. coli clinical isolates but not in other E. coli strains such as the non-pathogenic model strain BW25113 (22). Moreover, in silico analyses on speG in Shigella spp. predicted that all analyzed genome sequences harbored inactivating mutations within the ynfB-speG operon and a diverse panel of Shigella spp. exhibited over a twofold increase in endogenous spermidine when compared to E. coli (90). The increase in endogenous spermidine improved Shigella flexneri fitness in the adverse environment of murine macrophages (90). As such, it is conceivable that targeting SpeG would only be advantageous against bacteria with the intact enzyme. Additionally, targeting SpeG in backgrounds in which it is essential would be lethal with increased selective pressure for resistance development.
Similarly, polyamine analogs have been shown to sensitize K. pneumoniae and S. aureus to azithromycin (by up to 32-fold), mimicking the mechanism of natural polyamine-azithromycin synergy but at substantially lower concentrations (23, 25). Together, the effects of polyamines on bacterial responses to antibiotics provide a promising strategy to identify novel potential targets to combat AMR (Table 1). Nonetheless, the redundancy of polyamine biosynthesis pathways and the functional and, to some extent, structural homology between the bacterial and human enzymes in these pathways present challenges that will need to be addressed to selectively and effectively exploit these targets for antibacterial development. Further research into the identification of new inhibitors that are specific to targeting bacterial enzymes is needed to better quantify the therapeutic potential of targeting polyamine-related mechanisms.
PQS
PQS or 2-heptyl-3-hydroxy-4-quinolone is a quorum-sensing compound that mediates the production of numerous virulence factors and induces biofilm formation (118). PQS production is regulated by PqsR, a LysR-type transcriptional regulator that integrates PQS signaling into the broader quorum-sensing network, yet this molecule also exhibits non-quorum-sensing properties, which have been previously reviewed in reference 118. PQS controls the expression of 182 genes; 179 of those genes are controlled via PqsR-independent pathways, highlighting its extensive non-quorum-sensing role (119). Notably, sputum samples from cystic fibrosis patients infected with P. aeruginosa contain PQS in the nanogram/milliliter range, and clinical isolates from the sputum of young children exhibited a relative increase in PQS production compared to isolates taken from blood and urinary infections when cultured in a rich medium (120–122). These results suggest that PQS plays an important role in the long-term persistence of infection.
PQS alteration of antibiotic responses
PQS plays a role in bacterial antibiotic responses, where altering PQS levels, whether exogenously available or endogenously produced, has been shown to influence the antibiotic susceptibility of P. aeruginosa. Exogenous PQS sensitizes P. aeruginosa PAO1 to chloramphenicol, β-lactams, tetracycline, and aminoglycosides in disc diffusion assays (123). Furthermore, exogenous PQS and its endogenous overproduction by disrupting pqsL in PAO1 also increased susceptibility to fluoroquinolones (124). The loss of endogenous PQS by deletions in the PQS biosynthetic pathway (P. aeruginosa PAO1 ΔpqsH) results in increased resistance to aminoglycosides (by at least a twofold MIC shift) (125). A ΔpqsA mutant that exhibits loss of PQS production also exhibits increased tolerance, as determined by a higher CFU count after 3 hours of treatment, to fluoroquinolones and aminoglycosides (124). Additionally, a P. aeruginosa PAO1 small colony variant resistant to an aminoglycoside showed significant reduction in PQS production and mexGHI-opmD transcription (126). The deletion of mexI and opmD in PAO1 led to reduced endogenous PQS concentrations and resistance to fluoroquinolones and aminoglycosides in disk diffusion assays, similar to P. aeruginosa PAO1 ΔpqsH or ΔpqsA (123). Exogenous supplementation of PQS re-sensitized the ΔmexI and ΔopmD mutants to the antibiotics, as did the genetic complementation (123). These results further support PQS as a synergist of the above-mentioned antibiotics. However, additional studies were unable to reproduce the previously shown decrease in PQS production in mexGHI-opmD mutants of PAO1 and PA14 (127, 128). The discrepancies in these results may have arisen from condition-dependent effects (e.g., detection of PQS from liquid [123] vs solid [127] cultures). Nonetheless, P. aeruginosa appeared to lower its production of PQS in response to certain antibiotics, where microarray data of PAO1 showed that PQS biosynthesis genes (pqsA, pqsB, pqsC, pqsD, pqsE, and pqsH) are repressed upon exposure to sub-MIC concentrations of azithromycin, β-lactams, and fluoroquinolones (129). Conversely, P. aeruginosa PA14 treated with β-lactams exhibits no alteration in PQS production or accumulation (125). Both P. aeruginosa PA14 and PAO1 produce similar levels of PQS, but PAO1 PQS is mostly cell-associated (72%), whereas a majority of PA14 PQS (78%) is extracellular; thus, PA14 may not need to lower PQS production to mitigate PQS stress intracellularly (130). Notably, PQS may act as an endogenous stressor, evident from the ability of high levels of endogenously produced or exogenously added PQS to increase bacterial autolysis and sensitivity to antibiotics (123–125, 131).
Multiple mechanisms may underlie the ability of PQS to synergize with the above-mentioned antibiotics. One could be through the pro-oxidant activities of PQS (124, 132). It is probable, similar to polyamines, that PQS concentrations above their homeostatic range result in oxidative stress, increasing bacterial sensitivity to stressful conditions (124, 125, 133). Moreover, PQS sensitization to antibiotics could be through membrane alteration, reducing the membrane barrier effect for certain antibiotics. PA0011 in P. aeruginosa PAO1 encodes a 2-OH-lauroytransferase involved in lipid A biosynthesis and is negatively regulated by PQS (134). P. aeruginosa mutants with insertional inactivation of PA0011 exhibit a twofold or greater decrease in the MIC of fluoroquinolones, tetracycline, chloramphenicol, polymyxin, rifampicin, and β-lactams (134). P. aeruginosa strains deficient in PQS biosynthesis (ΔpqsR and ΔpqsH) exhibited increased PA0011 expression (134).
Conversely, PQS may improve the bacterial response to certain antibiotics. One example is that PQS production was also related to conferring carbapenem tolerance (135). Deletion of rpoN, an alternative sigma factor, upregulated pqsA, pqsH, and pqsR, increasing PQS production, and led to carbapenem tolerance in P. aeruginosa PAO1 (135). Exogenous PQS decreased bacterial killing by over 90% by the carbapenem biapenem in the P. aeruginosa wild-type strain, and the ΔrpoN ΔpqsA exhibited a loss of the carbapenem-tolerant phenotype, demonstrating that the carbapenem resistance phenotype in the ΔrpoN background is mediated by increased PQS (135). VqsR is involved in carbapenem tolerance in stationary phase P. aeruginosa cells, and vqsR knockout mutants exhibit higher bacterial cell death upon carbapenem treatment; the expression of VqsR is under the control of RpoN and PQS (136). Additionally, P. aeruginosa ΔptrA exhibits downregulation of PQS biosynthesis genes and a twofold decrease in the MIC of carbapenem (137).
The effect of PQS on susceptibility to other antibiotics, namely cationic antimicrobial peptides, is yet to be fully determined. The supernatant of subinhibitory colistin-treated P. aeruginosa PAO1 cultures shows increased PQS extracellular concentrations (138), yet P. aeruginosa PAO1 ΔpqsR mutants, which exhibit downregulated PQS synthesis genes, are PMB tolerant in bacterial time-kill curves (139).
PQS-mediated protection from antibiotics such as biapenem could be due, in part, to its effects on outer membrane vesicle (OMV) formation. PQS is trafficked between cells in OMVs, and the level of OMV formation is closely aligned with the level of PQS endogenously produced (130, 140–143). OMVs have been previously shown to sequester membrane-acting antimicrobials and carbapenems (144, 145), possibly contributing to increased resistance to these antibiotics, which would align with the observed increase in PQS synthesis by membrane-acting compounds (138). Interestingly, exogenous PQS is capable of forming OMVs not only in P. aeruginosa but also in E. coli and K. pneumoniae (142), suggesting its effects are not restricted to P. aeruginosa but extend to other bacterial species. These observations warrant further investigation into the effects of PQS on non-producers.
PQS plays a role in shaping bacterial populations in monoculture and polymicrobial communities. Interestingly, there was a concentration-dependent increase in PQS concentrations, detected by surface density, in aminoglycoside-treated P. aeruginosa PA14 swarming communities (125). PQS increased P. aeruginosa susceptibility to aminoglycosides as mentioned above. Yet, it was hypothesized that PQS may have an essential role in combating aminoglycoside stress by acting as a long-range life and death signal (124, 125, 146). The heightened PQS concentrations in response to aminoglycosides may serve as an attempt to prevent swarm expansion into the antibiotic-containing region, repelling approaching P. aeruginosa swarms far beyond that area (125, 146). This would suggest that PQS stress response is also a coordinator of spatial organization during swarming and is a collective stress response for the entire population (146). P. aeruginosa inoculated on semisolid agar in proximity to E. coli showed that P. aeruginosa steadily expanded toward E. coli and accelerated the production of PQS, which was detected by 48 h compared to 96 h in P. aeruginosa monoculture (147), suggesting that PQS has a spatiotemporal distribution that may garner an adaptive advantage for P. aeruginosa within its polymicrobial environment (147). PQS also has growth inhibitory activity against S. aureus and Bacillus subtilis, where total organic extracts from PA14 wild type but not a pqsA^–^ mutant inhibited the growth of both gram-positive species (148). PQS inhibited the motility of various gram-positive (e.g., B. subtilis, S. aureus, and Staphylococcus epidermidis) and gram-negative (e.g., E. coli and Proteus vulgaris) bacteria in semi-solid motility assays (149). Culture supernatants from several P. aeruginosa isolates stimulated biofilm formation in S. aureus, an effect positively correlated to the levels of PQS produced by the P. aeruginosa isolates and significantly reduced in PQS-deficient mutants (150). Together, these examples and others (reviewed in reference 118) show that PQS can lead to changes in biodiversity, richness, and structure of the microbial community. Together, PQS can both contribute to or respond to stress, depending on the context and type of antibiotic, and can help shape microbial communities, potentially conferring an advantage to P. aeruginosa in polymicrobial populations. Figure 2 summarizes the mechanisms of PQS-mediated alteration to antibiotic susceptibility.
Mechanisms of PQS-mediated alteration of antibiotic susceptibility. PQS synergizes with antibiotics by promoting oxidative stress and decreasing lipid A biosynthesis via downregulation of PA0011. Conversely, PQS could be antagonizing membrane-acting antibiotics by sequestering antibiotics with OMVs. PQS also impacts the surrounding bacterial population, where PQS secretion is upregulated in stressed P. aeruginosa populations to act as a long-range signal to repulse unstressed P. aeruginosa populations. Additionally, PQS can induce OMV production in other gram-negative bacteria, and the products from the pqsABCDH operon can inhibit gram-positive bacterial growth. Descriptions of each mechanism and the corresponding references are detailed in the text. Blue arrows denote increased, red arrows denote decreased, blunt-head arrows denote inhibited, dotted arrows denote the movement of a substrate, and open circled arrows denote synthesis.
Potential targets based on PQS alteration of antibiotic susceptibility
PQS has a complex role in shaping P. aeruginosa environments and in mediating stress responses that is not yet fully understood, but the elucidation of PQS mechanisms has shed light on multiple potential drug targets (Table 1). Interestingly, PQS itself has been shown to exhibit potential promise as an antibacterial, where PQS improved the survival of mice co-infected with S. aureus and P. aeruginosa (37). Potential analogs of PQS could work mechanistically similar to that of natural PQS, inducing antibiotic sensitivity, but at substantially lower concentrations, similar to the strategy employing polyamine analogs described above (25). Although PQS was shown to increase P. aeruginosa susceptibility to tobramycin, it was overexpressed in response to tobramycin treatment, suggesting PQS may serve an important role for survival of the population. As such, PqsR is a potential target due to its positive feedback effect on PQS production. Quinazoline disulfide analogs, quinazoline analogs, 3-hydroxy-pyridinone analogs, and other PqsR inhibitors (see Table 1) have been shown to potentiate tobramycin in P. aeruginosa-infected murine models, lowering cell viability of both S. aureus and P. aeruginosa in a mixed biofilm and of P. aeruginosa biofilms (26–30). Although exogenously supplemented PQS was shown to confer antibiotic susceptibility to fluoroquinolones, aminoglycosides, and tetracyclines, inhibitors of PqsR and PqsR/PqsD synergized with fluoroquinolones and aminoglycosides against planktonic cells, with fluoroquinolones and tetracycline against P. aeruginosa biofilms, and with aminoglycosides against P. aeruginosa in Caenorhabditis elegans infection assays (Table 1) (26, 28, 31–33, 35). Moreover, targeting PQS biosynthesis could lower PQS-induced protection to carbapenems and polymyxin. PqsA inhibitors norharmane and a 3-hydroxy-pyridinone analog each synergized with polymyxin to treat P. aeruginosa planktonic cells and a norharmane-polymyxin combination yielded lower bacterial burden in a murine lung infection model (Table 1) (28, 36). Additionally, norharmane and a dual PqsR/PqsBC inhibitor (a benzamide-benzimidazole analog) each synergized with carbapenem to treat P. aeruginosa planktonic cells (34, 36).
PAA
PAA is an intermediate by-product in the degradation of aromatic amino acids, yielding the common metabolic intermediates succinyl-CoA and acetyl-CoA (151). The operon paaABCDEFGHJKXYI encodes the enzymatic pathway for PAA catabolism and is one of the most differentially regulated pathways in bacteria in response to environmental stimuli (152–156). PAA is a known signaling compound in plants, acting similarly to an auxin, regulating plant growth and development (157); auxins are known to have regulatory effects on surrounding soil bacteria (158). PAA is also detected in humans and is predominantly derived from gut microbiota-dependent metabolic conversions (159); hence, bacteria may encounter PAA during infection. PAA and its catabolism have been implicated in bacterial pathogenesis in several infection models (38, 41, 160–162). In an A. baumannii zebrafish infection model, PAA injection into zebrafish otic vesicles (as little as 3.4 ng) or increased PAA excretion due to the loss of bacterial PAA catabolism via ΔpaaA or ΔgacS (positive regulator of the paa operon) enhanced neutrophil migration and clustering at the site of infection (39, 160), ultimately leading to greater bacterial killing and improved host survival (160). Blocking PAA catabolism by deleting paaB also resulted in attenuated virulence of A. baumannii in a murine catheter-associated urinary tract infection model (41). Similarly, PAA catabolism was required for the pathogenicity of B. cenocepacia in C. elegans (38), whereas P. aeruginosa-infected C. elegans exhibited increased survival upon 200 µg/mL PAA treatment (161). However, C. elegans infected with S. aureus, S. epidermidis (PAA non-catabolizing pathogens), and B. cenocepacia did not have altered survival rates upon PAA treatment, but a B. cenocepacia ΔpaaABCDE mutant treated with PAA did exhibit increased C. elegans survival (162). Furthermore, genes specifically associated with dominant clonal clusters of Mycobacteroides abscessus infecting cystic fibrosis patients were enriched for PAA catabolism, pointing to the potential infection relevance of PAA (163). Together, PAA acts as a regulatory signal that bacteria may encounter during infection, which could contribute to establishing persistent infections and responding to antibiotic stress.
PAA alteration of antibiotic responses
PAA catabolism may contribute to bacterial response to antibiotic and oxidative stress. The paa operon is highly expressed in A. baumannii persister cells induced by aminoglycosides, β-lactams, PMB, tetracycline, and fluoroquinolones (154, 164). Likewise, A. baumannii upregulates the paa operon upon treatment with trimethoprim-sulfamethoxazole (41). Knockouts of paaB in A. baumannii Ab17978 and the pathogenic UPAB1 strains have increased endogenous levels of PAA, coinciding with a twofold decrease in ciprofloxacin, erythromycin, and zeocin MIC in both strains, with the latter exhibiting a 2-log reduction in bacterial survival when exposed to H_2_O_2_ relative to the parent strains (41). Interestingly, A. baumannii UPAB1 ΔpaaB exhibits a fourfold decrease in aminoglycoside MIC, while A. baumannii Ab17978 ΔpaaB, whose wild type is already fourfold more susceptible to aminoglycosides than UPAB1, exhibits no further change in MIC but does exhibit a reduction in growth under aminoglycoside treatment (41). Moreover, PAA degradation played a role in spontaneous resistance to carbapenems in B. cenocepacia (165). The mechanisms of PAA-related alteration to antibiotic susceptibility are summarized in Fig. 3.
Mechanisms of PAA-related alteration of antibiotic susceptibility. In gram-negative bacteria, promoting PAA accumulation, through either the inhibition of catabolism or uptake of exogenous PAA, leads to increased sensitivity to aminoglycosides and H2O2 and may decrease virulence. PAA degradation has been shown to be positively regulated by antibiotic exposure, and PAA plays a role in carbapenem resistance and persister cell formation. For detailed descriptions of each mechanism and the corresponding references, refer to the text. Blue arrows denote increased, dotted arrows denote the movement of a substrate, and open circled arrows denote catabolism.
Potential targets from PAA alteration of antibiotic susceptibility
To date, there are no known inhibitors that have been directly confirmed to target proteins encoded by the paa operon. However, targeting the proteins early in the PAA metabolic pathway may serve as viable drug targets to enhance susceptibility to aminoglycoside (and possibly other antibiotics mentioned above) and increase bacterial killing through neutrophil recruitment (Table 1) (41, 160). Analogs of PAA could potentially serve as antibiotic adjuvants, acting similarly to the natural PAA. The PAA analog diclofenac, a non-steroidal anti-inflammatory, has antimicrobial activity and potentiates streptomycin, an aminoglycoside, against E. coli and S. aureus in checkerboard assays, potentiates colistin against K. pneumoniae, P. aeruginosa, and A. baumannii in vitro and against A. baumannii in vivo in a murine model of pneumonia (166, 167). The increased aminoglycoside sensitivity was shown to be a result of inhibition of bacterial DNA synthesis by diclofenac, and the increased colistin sensitivity was largely attributed to a decrease in type IV pili, but repression of PAA catabolism was also observed (166, 167). Additionally, targeting PAA catabolic enzymes could attenuate virulence; the phenotypes of paa gene deletion mutant phenotypes of B. cenocepacia, A. baumannii, and P. aeruginosa in in vivo models provide proof-of-concept for such an approach (38–41, 161). The role of PAA as a signaling molecule within the microbial community, its impact on antibiotic susceptibility, and the identification of inhibitors require further research.
Indole
Indole is an aromatic heterocyclic organic compound produced via the tryptophanase TnaA from tryptophan by multiple gram-negative and gram-positive bacteria and was shown to influence antibiotic resistance and persistence (168–170). The physiologically relevant concentration of indole varies across the bacterial growth cycles; for example, E. coli cells produce indole bursts (>60 mM) when entering the stationary phase, with indole concentrations subsiding soon after (171); this form of indole signaling is termed pulse signaling and is only experienced by the producer cell (172). The growth medium of E. coli grown into the stationary phase contains approximately 0.5–1.0 mM, whereas in vivo concentrations vary across body sites and individuals, where human stool samples range from 0.3 to 6.64 mM and plasma concentrations from 5.12 to 72.6 nM (172–174). These indole concentration ranges are typically considered physiologically relevant for long-term exposure; such persistent signaling can be experienced by producer and non-producer cells (172).
Indole alteration of antibiotic responses
Indole has been both shown to inhibit and promote persister formation, depending on the experimental design, including the antibiotic-indole combinations and concentrations and timing of indole exposure, as reviewed by Zarkan et al. (172). New research has since provided evidence of indole pulse signaling being responsible for the formation of quinolone E. coli persisters (171, 175). An artificial pulse of indole in quinolone-treated E. coli ΔtnaA (indole-negative) led to an increase in quinolone persister cells. In the absence of exogenous indole, this mutant exhibited a 2–10-fold decrease in quinolone persister formation compared to the wild type (175). Quinolones inhibit DNA gyrase binding to gyrase A; indole binds to gyrase B, an interaction hypothesized to protect against quinolones while reducing DNA gyrase activity, inhibiting DNA replication, and inducing a dormant state (175, 176). Notably, indole appeared to confer protection against fluoroquinolone in vivo, where C. elegans co-infected with E. coli ΔtnaA and S. Typhimurium in an intestine infection model had a lower S. Typhimurium bacterial load compared to a co-infection with indole-producing wild-type E. coli after fluoroquinolone treatment (42). Indole does not have an effect on novobiocin persister formation, an expected result given that they both interact with gyrase B (175). Moreover, indole is produced by Vibrio cholerae in response to sub-MIC aminoglycosides, and the exogenous addition of indole during near-exponential phase culture increases persistence to aminoglycosides (177). Indole induces the expression of the ribosome-associated factor RaiA*,* which is proposed to result in the preservation of non-translating inactive but intact ribosomes that could be rapidly reactivated upon stress relief, conferring a survival advantage (177). Interestingly, indole does not have an effect on V. cholerae persistence to carbenicillin or trimethoprim (177). In contrast, E. coli ΔtnaA showed an increase in ampicillin (β-lactam) persisters during the exponential phase relative to the wild type; however, the opposite was observed during the stationary phase (178). E. coli persisters that survive ampicillin treatment display a lower intracellular pH and a tighter grouping of pH within the persister population (178). Conversely, E. coli ΔtnaA exhibits a higher cytoplasmic pH, and artificial pulsing restores the pH to that of the wild type (178, 179). These examples suggest that the effects of indole on persister formation may be antibiotic and growth-phase-specific. However, some of the disparities in the reported effects of indole on bacteria may have stemmed from effects related to different solvents used to solubilize indole (e.g., DMSO vs ethanol; reviewed by Song and Wood [180]). Furthermore, potential discrepancies between expected outcomes of bacterial exposure to exogenous indole and the phenotypes of a ∆tnaA strain may stem from the fact that the mutant not only lacks endogenous indole production but also accumulates tryptophan and lacks Tna-dependent pyruvate production, which together may confound the indole-related results. These variables should also be considered when interpreting the findings and comparing the different studies.
In addition to its role in persister formation, indole alters the susceptibility of multiple gram-negative bacteria to antibiotics. Indole is overproduced during sub-MIC aminoglycoside exposure in V. cholerae and is secreted by E. coli spontaneous drug-resistant mutants, shielding sensitive strains from antibiotic insult (177, 181). Additionally, exogenous indole increases antibiotic resistance in E. coli, K. pneumoniae, P. aeruginosa, and S. Typhimurium as determined in vitro by CFU counting and disk diffusion assays (42, 182–185). Indole-mediated alterations of antibiotic susceptibility are associated with its effects on efflux pump activation; these have been previously reviewed (172), hence will not be detailed herein. Conversely, indole from donor E. coli extracts was more recently shown to sensitize P. aeruginosa, Proteus mirabilis, and K. pneumoniae clinical isolates to fluoroquinolones, β-lactams, and aminoglycosides (186). qRT-PCR analyses concluded that indole downregulated genes encoding RND-type efflux in K. pneumoniae (186). On the other hand, indole has recently been shown to facilitate the uptake of the antimicrobial quaternary ammonium cation, malachite green, in E. coli and P. aeruginosa (187). Time-resolved second-harmonic light scattering showed that micromolar concentrations of indole increased the rate of MG transportation across the bacterial cytoplasmic membrane via an Mtr permease and enhanced passive diffusion (187).
Interestingly, indole signaling may overlap with PQS signaling in P. aeruginosa. Indole was shown to induce P. aeruginosa PAO1 resistance to tetracycline, aminoglycosides, and β-lactams in vitro by repression of mexGHI-opmD and PQS synthesis genes (185). Indole supplementation does not further decrease PQS concentrations in a pqsR knockout mutant, suggesting that indole signaling is linked to PQS via pqsR suppression (185). Resistance to aminoglycosides and tetracycline matches previous reports discussed under the PQS alteration of antibiotic susceptibility section (123, 125, 185). The mechanisms of indole-mediated alteration to antibiotic susceptibility are summarized in Fig. 4.
Mechanisms of indole-mediated alteration of antibiotic susceptibility. Indole has two forms of signaling, a low-level consistent signaling (persistent signaling) where bacteria are consistently exposed to 0.005–6.64 mM of indole, and pulse signaling, in which bacteria are temporarily exposed to indole concentrations of up to 60 mM. Low-level consistent signaling can synergize antibiotics by downregulating RND-type efflux pumps and has been shown to promote malachite green uptake, increasing transportation across the membrane. Conversely, low-level consistent signaling can antagonize antibiotics by activating efflux pumps and inhibiting PQS synthesis in P. aeruginosa and promote persister cell formation in V. cholerae via inducing the expression of RaiA. Indole has also been shown to be secreted from aminoglycoside-resistant sub-populations of E. coli and protect some of the surrounding sensitive population. Indole pulse signaling has been shown to inhibit GyrB and promote quinolone persister cell formation and to be important for the maintenance of cytoplasmic pH. For detailed descriptions of each mechanism and the corresponding references, refer to the text. Blue arrows denote increased, red arrows denote decreased, blunt-head arrows denote inhibited, dotted arrows denote the movement of a substrate, black flat-headed arrows denote inhibition, and open circled arrows denote synthesis.
Potential targets based on indole alteration of antibiotic susceptibility
Indole appears to be mediating variable effects on bacterial responses to antibiotics, and more conclusive results may be needed; nonetheless, TnaA represents a viable antibiotic adjuvant drug target to mitigate indole-mediated antibiotic resistance and persister formation. Multiple TnaA inhibitors have been identified, such as ALG-05, and shown to reduce cecal indole and serum indoxyl sulfate, a product of microbial tryptophanase, in vivo and inhibit indole production by E. coli in vitro (Table 1) (43–45). However, these compounds have yet to be tested in combination with antibiotics. Furthermore, indole derivatives have been shown to eradicate E. coli, S. aureus, P. aeruginosa, S. epidermidis, Enterobacter tabaci, and Mycobacterium tuberculosis persister cells alone and in combination with antibiotics (recently reviewed in reference 180).
Central carbon and nitrogen metabolism-linked metabolites
Due to the high interconnection of metabolic pathways, the environmental scarcity or abundance of metabolites can alter the metabolic states of bacteria, whereby key metabolites can activate slowed metabolisms, which is a characteristic of antibiotic-tolerant and persister cells (13, 188). Hence, the abundance of host- or bacterial-derived metabolite intermediates from the central carbon and nitrogen metabolism, including but not limited to pyruvate, succinate, glutamine, and alanine (see Table 1 for the full list), can activate metabolisms, promoting bacterial killing by bactericidal antibiotics (recently reviewed in reference 13). Similarly, the host immunometabolite itaconate, which is derived from the tricarboxylic acid cycle intermediate cis-aconitate, can be used as a carbon source by P. aeruginosa (189, 190). Additionally, itaconate has been shown to alter P. aeruginosa virulence and antibiotic susceptibility (190–192). Host itaconate, glutamate, and succinate have been shown in ex vivo and in vivo models to be secreted at high enough concentrations to alter antibiotic susceptibility and virulence (190–193). This topic has been recently reviewed in depth by Peng et al. (13) through the context of metabolic reprogramming; hence, this section will provide only select examples of the effects of central carbon and nitrogen metabolism-linked metabolites on antibiotic susceptibility.
Central carbon and nitrogen metabolism-linked metabolite-mediated alteration of antibiotic responses
Allison and colleagues (46) showed that supplementing carbon sources that enter upper glycolysis, as well as pyruvate, with aminoglycosides increased bacterial killing of E. coli and S. aureus persister cells. These metabolites activate central metabolism and the electron transport chain, which elevates the bacterial proton motive force (PMF), increasing aminoglycoside uptake (46). Similar observations were reported with aminoglycoside-resistant Edwardsella tarda, Salmonella spp., P. aeruginosa, K. pneumoniae, Vibrio alginolyticus, and Vibrio parahaemolyticus (47–54). Similarly, pyruvate, inosine, and fructose-driven increases in PMF have been shown to promote tetracycline uptake in tetracycline-resistant E. coli and K. pneumoniae, ampicillin uptake in Streptococcus agalactiae, and colistin lipid A binding in colistin-resistant V. alginolyticus, E. coli, E. tarda, and K. pneumoniae (55–57). Furthermore, metabolite supplementation can improve antibiotic uptake by enhancing porin expression; for example, glutamine and inosine increase β-lactam killing in E. coli by upregulating the OmpF porin, and inosine increases tetracycline killing of K. pneumoniae by upregulating the OmpK 36 porin (55, 58).
Interestingly, itaconate improves P. aeruginosa resistance toward aminoglycosides by an undescribed mechanism but can inhibit bacterial growth in other gram-negative and gram-positive bacteria, most of which are unable to catabolize itaconate (194–196). Itaconate inhibits the glyoxylate cycle enzyme isocitrate lyase in E. coli and S. Typhimurium and slows energy metabolism in S. aureus, which consequently induces aminoglycoside tolerance in S. aureus (194, 195, 197).
Certain metabolites can also promote ROS accumulation. Pyruvate potentiates aminoglycoside lethality against E. tarda, P. aeruginosa, E. coli, K. pneumoniae, and methicillin-resistant S. aureus by boosting the pyruvate-cysteine-GSH system/glycine-ROS metabolic pathway, which results in ROS generation (59). Nitrite activates the pyruvate cycle and electron transport chain, inducing ROS generation while repressing antioxidants in P. aeruginosa, potentiating cefoperazone-sulbactam against P. aeruginosa resistant to that combination (60). Together, these findings highlight that metabolites in high abundance in the bacterial environment can alter cell survival by altering antibiotic uptake and increasing ROS.
Potential strategies based on central carbon and nitrogen metabolism-linked metabolite-mediated alteration of antibiotic responses
Co-administering antibiotics with key metabolites to reprogram the metabolism of antibiotic-resistant, tolerant, or persister bacteria into susceptible states is a promising strategy to improve the therapeutic outcome of current antibiotics. Aminoglycosides combined with metabolite intermediates from the central carbon metabolism have reduced the bacterial load and improved host survival in mouse, zebrafish, and Huiyang bearded chicken infection models infected with V. alginolyticus, Salmonella spp., or E. tarda (13, 47–51, 53). The combination of pyruvate with colistin to treat colistin-resistant V. alginolyticus in a zebrafish infection model yielded a higher survival rate when compared to the colistin-treated group (56). Ampicillin combined with fructose or glutamine improved the survival of zebrafish infected with ampicillin-resistant S. agalactiae and increased the survival of mice infected with E. coli, P. aeruginosa, or K. pneumoniae while also reducing the bacterial load in blood, liver, and spleen (57, 58). Inosine-tetracycline combination improved mouse survival by fivefold in a peritonitis-sepsis infection model and resulted in a lower bacterial load in the heart, lung, and liver when compared to tetracycline treatment (55). These data support the potential use of this strategy to combat antibiotic resistance and persister infections. Notably, the metabolites achieved in vivo effectiveness at concentrations well below previously reported tolerable amounts in mammals in some of the studies reported herein (47, 58, 198, 199), suggesting that this strategy could provide a safe and effective potential therapeutic application.
Microbial secreted volatile-mediated communication of resistance
In recent years, there has been an emerging interest in complex volatile compounds playing a role in bacterial interactions (200). Volatile compounds constitute a large class of infochemicals (chemicals involved in signaling, communication, and transmission of information between cells) characterized by their high vapor pressure, low boiling point, and low molar mass (201, 202). A database of identified microbial secreted VOCs has been published (201). This section will review VOCs relevant to human bacterial infection shown to alter antibiotic resistance, tolerance, or persistence, namely nitric oxide (NO), hydrogen sulfide (H_2_S), ammonia, 2,3-butanedione, and trimethylamine (TMA) (Fig. 5).
Mechanisms of VOC-mediated alteration of antibiotic susceptibility. H2S-mediated antibiotic resistance is attributed to H2S-mediated increase in antioxidant capacity and the oxidation of H2S into sulfane sulfur, which de-represses antibiotic efflux pumps. H2S can also react with cysteine to produce cysteine hydropersulfide, which degrades β-lactams into inactive forms. NO potentiated H2O2 killing by increasing double-strand DNA breaks and decreasing GSH concentrations. Conversely, NO can protect bacteria from H2O2 by increasing the antioxidant capacity of cells. It can also protect bacteria from β-lactams. The loss of bacterial nitric oxide synthase attenuates virulence in macrophages. Both ammonia and TMA protect gram-negative and gram-positive bacteria from tetracycline, whereby, in E. coli, ammonia promotes polyamine synthesis, increasing the antioxidant capacity of the cell, and TMA increases cytoplasmic pH, reducing tetracycline uptake. 2,3-butanedione induces persister formation in E. coli upon β-lactam and tetracycline exposure, potentially by regulating ypdB. For detailed descriptions of each mechanism and the corresponding references, refer to the text. Blue arrows denote increased, red arrows denote decreased, blunt-head arrows denote inhibited, dotted arrows denote the movement of a substrate, black flat-headed arrows denote inhibition, open circled arrows denote synthesis, and the blue membrane denotes both a gram-negative and gram-positive membrane.
Hydrogen sulfide alteration of antibiotic responses
Loss of H_2_S production through chemical or genetic inactivation of cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST) resulted in increased in vitro susceptibility of the pathogens Bacillus anthracis, P. aeruginosa, S. aureus, and E. coli to aminoglycosides, β-lactams, fluoroquinolones, and H_2_O_2_, which was chemically complemented by exogenous H_2_S (61, 63, 203). Of note, genetic perturbation of H_2_S production in P. aeruginosa PAO1 by deletion or overexpression in another study yielded no change in antibiotic susceptibility (204). Chemical inactivation of bacterial CSE, CBS, and 3-MST was achieved by DL-propargylglycine (PAG), amino-oxyacetate (AOAA), and aspartate (Asp), respectively (61, 203). In contrast, Weikum and colleagues (205) reported that exogenous H_2_S synergized with tetracyclines, quinolones, and β-lactams against S. aureus in disk diffusion assays; they only observed sulfide-mediated protection from aminoglycosides, yet the inhibitor AOAA did not synergize with gentamicin in checkerboard assays, nor did gentamicin induce H_2_S production. Previously, PAG and AOAA were both combined with gentamicin to show synergy against S. aureus in bacterial killing experiments (203). The conflicting findings are likely due to the type of assays performed and strain variations, especially since S. aureus HG003, tested by Weikum et al. (205), did not exhibit substantial intracellular H_2_S levels. Interestingly, intracellular H_2_S production levels of several E. coli urinary tract infection clinical isolates positively correlated with their extents of antibiotic resistance (61). Conversely, H_2_S levels were lower in resistant and multidrug-resistant P. aeruginosa clinical isolates from cystic fibrosis patients relative to sensitive isolates (204). Together, the effects of H_2_S on antibiotic susceptibility may not be universal.
Various mechanisms contribute to H_2_S-mediated antibiotic resistance. Treatment with either macrolides or H_2_O_2_ induced H_2_S synthesis, and in turn, H_2_S stimulated the activities of catalase and superoxide dismutase and inhibited the Fenton reaction, which generates ROS (203). Additionally, H_2_S delayed killing of E. coli cells by antibiotics in bacterial in vitro time-kill analyses by downregulating cytochrome bo oxidase and inducing cytochrome bd oxidase I/II to maintain respiratory flux and redox balance and bolster the bacterial antioxidant capacity (61). The oxidation of H_2_S produces sulfane sulfur, which has been shown to react with regulatory thiols of the transcriptional regulator MexR in P. aeruginosa, leading to de-repression of MexAB-OprM efflux pumps, which increases antibiotic resistance (206). H_2_S can also react with cysteine to produce cysteine hydropersulfide, which can decompose β-lactams into their inactive form, contributing to intrinsic bacterial resistance to β-lactams (207).
Nitric oxide alteration of antibiotic responses
Nitric oxide is a soluble gas that is produced at the infection site by both the host macrophage cytokine-inducible nitric oxide synthase (iNOS) and the infecting microbe bacterial nitric oxide synthase (bNOS) (208, 209). Macrophages and neutrophils kill ingested pathogens using a multitude of factors, which include H_2_O_2_ oxidative stress (208). During infection, excess NO is produced by iNOS; iNOS-deficient mice display increased bacterial growth and susceptibility to infection compared to wild-type mice, suggesting NO has a role in host innate resistance to infection (210).
NO both synergized and antagonized H_2_O_2_ against different bacteria. Exogenous NO has been shown to potentiate H_2_O_2_ killing of E. coli (211–213). E. coli treated with the NO donor diethylamine/NO exhibited no cytotoxicity, but the addition of H_2_O_2_ led to a 10,000-fold increase in bactericidal effects (211). The synergistic combination of NO and H_2_O_2_ led to increased double-strand DNA breaks, altered cellular respiration, and decreased antioxidant GSH concentration, and indirect evidence suggested metal ions played a key role in the NO/H_2_O_2_ bactericidal mechanism (211, 213). Conversely, 30 µM NO increased the resistance of B. subtilis to 10 mM H_2_O_2_ (lethal dose) by 100-fold (214). The protection was hypothesized to be through a dual mechanism, whereby NO boosts the activity of pre-existing H_2_O_2_ scavenging enzymes and inhibits the Fenton reaction by transiently interrupting cysteine reduction by inhibiting Trx/TrxRed (214). Interestingly, this mechanism is similar to that of H_2_S, and NO-deficient B. anthracis Δnos cells had increased H_2_S production, suggesting that both systems share a role in bacterial response to ROS (203). Moreover, B. anthracis uses bNOS-generated NO to combat host immune oxidative stress, and the loss of bNOS greatly attenuates virulence (215).
Several bacteria produce NO endogenously via bNOS (216). B. subtilis ΔbNOS exhibits reduced bacterial growth compared to the wild type when challenged with β-lactam, aminoglycoside, and quinolone antibiotics, and exogenous 100 µM NO temporarily protects the cells from β-lactam toxicity (217). S. aureus ΔbNOS was also more susceptible to β-lactams in turbidimetric and CFU counting time-dependent assays (217). It was also shown indirectly that treatment of β-lactams in B. subtilis stimulates bNOS activity (217). The mechanisms of NO-mediated alteration of antibiotic susceptibility are yet to be fully explored.
Ammonia and TMA alteration of antibiotic responses
Placing E. coli spent media in one compartment next to bacteria cultured in another compartment in two-petri-dish assays led to protection of E. coli, P. aeruginosa, B. subtilis, and S. aureus from tetracycline due to aerial exposure to volatile compounds produced by E. coli (110, 218). Testing pure volatile compounds known to be released from E. coli revealed that ammonia and TMA both induced tetracycline resistance (110, 218). Spent media of E. coli with disrupted ammonia production (ΔaspC, ΔpurA, or ΔaspA) did not aerially induce tetracycline resistance (110). Aerial ammonia increased the intracellular pool of polyamines, and impairment of polyamine biosynthesis significantly reduced the protective effects of aerial ammonia (110). In addition, ammonia could combat oxidative stress induced by paraquat, similar to polyamines (110). Taken together, ammonia-induced tetracycline resistance appears to be a result of polyamine-induced protection (110).
To show the effects of TMA independent of aerial ammonia, an aspC E. coli mutant was used in the presence of TMA N-oxide (TMAO, a TMA substrate) as the VOC donor protected E. coli, P. aeruginosa, B. subtilis, and S. aureus from tetracycline and chloramphenicol; tetracycline resistance was lost upon disruption of TMAO to TMA conversion (218). The effects of TMA were not dependent on polyamine biosynthesis and did not reduce paraquat oxidative stress (218). The resulting tetracycline protection was shown to be a result of increased pH, causing a reduction in tetracycline uptake (218).
2,3-butanedione alteration of antibiotic responses
The VOC 2,3-butanedione, emitted by S. aureus, E. coli, and K. pneumoniae, has been shown to induce E. coli persister cell formation upon treatment with β-lactam and tetracycline antibiotics (219–221). The dominant factor for 2,3-butanedione persister formation is the induction of hipA and hipB genes; hipA overexpression is a known mediator of persister formation (221). Furthermore, the exposure of E. coli to 2,3-butanedione downregulated all 30 genes associated with chemotaxis and motility (221). Interestingly, an E. coli ypdB knockout mutant exposed to 2,3-butanedione exhibited no change in swarming motility and persister cell formation, suggesting it may be a regulator of 2,3-butanedione effects in E. coli (221).
Potential targets from VOC alteration of antibiotic susceptibility
Leveraging VOC-related phenotypes may provide new potential antimicrobial strategies. Aspartate, a known inhibitor of 3-MST, suppressed the growth of E. coli in the presence of ampicillin (61). Inhibitors of 3-MST, CBS, and CSE enzymes showed enhanced in vitro clearance of S. aureus by leukocytes (62). CSE is the primary source of H_2_S production in S. aureus and P. aeruginosa, and CSE inhibitors in these pathogens have been shown to potentiate aminoglycosides in both in vitro and in a murine infection model (Table 1) (63). Additionally, the genetic or chemical inactivation of CSE lowers persister viability upon fluoroquinolone treatment (63). A double deletion of cbs and cse in S. aureus and deletion of the 3-MST homolog in E. coli (ΔsseA) exhibited potentiated host rapid immune-mediated killing and improved bacterial clearance in severe burn models (62). The drug delivery vector Gm@UiO-66-MA, which sequesters bacterially produced H_2_S, enhances the susceptibility of tolerant E. coli bacteria to aminoglycosides in in vitro and in vivo models (64). Additionally, a compound called 7B that scavenges H_2_S increases bacterial killing of E. coli, S. aureus, and P. aeruginosa when challenged with gentamicin and ciprofloxacin (65). 7B also marginally boosts bacterial clearance from macrophages and polymorphonuclear neutrophils and synergizes with gentamicin in a P. aeruginosa-infected pneumonia mouse model (65).
There are no bNOS inhibitors known to date, but NO-releasing polymeric implant devices potentiated various antibiotics against S. aureus, P. aeruginosa, E. coli, B. subtilis, Enterococcus faecalis, Haemophilus influenzae, and A. baumannii biofilms (reviewed extensively in references 66–68). On the other hand, PurA may serve as a target for inhibition of ammonia production; aurodox has been recently shown to inhibit PurA but exhibits insufficient target selectivity (69). PurA has also recently been recognized as a potential adjuvant drug target for colistin in E. coli (70). Furthermore, targeting YpdB, the potential master regulator of 2,3-butanedione effects on E. coli, may serve as a potential adjuvant target that has not yet been explored.
LIMITATIONS, FUTURE DIRECTIONS, AND CONCLUDING REMARKS
Chemical-mediated alteration of antibiotic resistance is an emerging field in which discoveries may elucidate novel drug targets. AMR is an accelerating problem, compounded by the inherent finite number of druggable targets within a biological system; hence, exploring and exploiting all potential targets and therapeutic strategies is needed to combat infection. The concept of adjuvant compounds that inhibit bacterial resistance mechanisms provides an appealing strategy to revitalize current treatment regimens. Given the advances reviewed herein in our understanding of the role signaling compounds play in antibiotic resistance, tolerance, and persister formation, these small molecular cues offer a promising avenue for new drug targets.
Despite recent progress in this emerging field, several areas remain in need of clarification. For example, the field would benefit from clarification of the effective concentrations of these small molecular cues in the bacterial microenvironment at the infection site, which would better guide appropriate concentrations for in vitro assays. The concentration of these compounds may be body site-dependent, and a fraction of some chemicals, such as polyamines, may be bound to macromolecules, possibly complicating the determination of the free-molecule concentration. Nonetheless, in vitro phenotypes of some chemicals described in this review, tested at concentrations within their in vivo concentration ranges, have been validated in in vivo assays. Together, accurately identifying in vivo levels of the various small molecules and establishing in vivo models with adequate controls to validate in vitro phenotypes will improve clarity related to the effects of small molecules on antibiotic therapeutic outcomes.
Another limitation to our current understanding of chemically altered antibiotic susceptibility phenotypes is that they are mostly derived from lab-adapted reference strains with limited data from clinical isolates or diverse bacterial strains. As such, some strain-specific phenotypes may be erroneously generalized, and chemical-mediated antibiotic phenotypes and molecular mechanisms stemming from differences in regulatory components or other structures across bacteria may be overlooked. Therefore, it is advisable to validate the phenotypes of interest and their mechanisms across diverse sets of bacterial isolates.
Many of the potential targets described above require further validation and characterization; the inhibitors of such targets need testing to ensure in vivo efficacy and limited off-target effects. Additionally, the potential for compounding effects of multiple chemicals at the infection site, as previously shown with polyamines and bicarbonate (25), could be missed if the effects of one compound are minimal or dependent on the presence of another. Notably, the potential cumulative effects of chemicals at the site of infection on bacterial responses to antibiotics could range from synergistic (potentially leading to drastic antibiotic susceptibility shifts) to antagonistic (canceling the effects of one another). These possibilities warrant in-depth assessments of the combinatorial effects of host chemicals and others that bacteria encounter at the site of infection. The often complex and variable nature of phenotypes across bacterial type, growth stage, polymicrobial environment composition, and infection location can make targeting chemical-mediated effects challenging. It is plausible that inhibiting one form of chemical-mediated resistance, which is used as a chemical signal in one organism, could create a niche for another organism to take over.
Several additional questions remain in this area of research, including
What other small molecules, secreted by bacteria or the host that bacteria encounter during infection, can modulate antibiotic resistance? What is the influence of exposure to such small molecules on antibiotic therapeutic outcomes in vivo?What are the exact molecular mechanisms by which the small molecules exert their effects on antibiotic resistance?How do bacteria sense and respond to these chemicals in the context of antibiotic susceptibility? What are the regulatory cascades that link the uptake, biosynthesis, efflux, and catabolism of these molecules to the antibiotic response?What are the combined effects of the different chemicals that might co-exist at the site of infection on bacterial response to antibiotics?Is there a hierarchy in the response of different bacteria to these signals that might influence the potential cumulative effects of chemicals at the site of infection and the development of a survival niche for specific organisms and not others?
Addressing these questions and clarifying the limitations outlined above will further enhance our understanding of the chemical-mediated alterations in antibiotic response and their potential influence on antibiotic therapeutic outcomes. Together, elucidating the mechanisms of chemical-mediated resistance in bacteria is a largely overlooked avenue that may offer new, much-needed therapeutic strategies to curb the rise of AMR and combat bacterial infections.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Public Health Agency of Canada. 2023. Pan-canadian action plan on antimicrobial resistance. Available from: https://www.canada.ca/en/public-health/services/publications/drugs-health-products/pan-canadian-action-plan-antimicrobial-resistance.html
- 2World Health Organization. 2019. Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline. Genrva World Health Organization
- 3Centers for Disease Control and Prevention. 2022. Antimicrobial resistance threats in the United States, 2021-2022. CDC, Atlanta, GA Department of Health and Human Services
- 4Naghavi M, Vollset SE, Ikuta KS, Swetschinski LR, Gray AP, Wool EE, Robles Aguilar G, Mestrovic T, Smith G, Han C, et al.. 2024. Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet 404:1199–1226. doi:10.1016/S 0140-6736(24)01867-139299261 PMC 11718157 · doi ↗ · pubmed ↗
- 5Nikaido H. 2001. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol 12:215–223. doi:10.1006/scdb.2000.024711428914 · doi ↗ · pubmed ↗
- 6Dhanda G, Acharya Y, Haldar J. 2023. Antibiotic adjuvants: a versatile approach to combat antibiotic resistance. ACS Omega 8:10757–10783. doi:10.1021/acsomega.3c 0031237008128 PMC 10061514 · doi ↗ · pubmed ↗
- 7Wright GD. 2016. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol 24:862–871. doi:10.1016/j.tim.2016.06.00927430191 · doi ↗ · pubmed ↗
- 8El-Halfawy OM, Valvano MA. 2012. Non-genetic mechanisms communicating antibiotic resistance: rethinking strategies for antimicrobial drug design. Expert Opin Drug Discov 7:923–933. doi:10.1517/17460441.2012.71251222860901 · doi ↗ · pubmed ↗