The staphylococcal type VII secretion system protein EsxC impacts daptomycin sensitivity through controlling bacterial cell envelope integrity
Victoria Smith, Robeena Farzand, Giridhar Chandrasekharan, Kate E. Watkins, Ramon Garcia Marset, Jeannifer Yap, Sebastien Perrier, Arnaud Kengmo Tchoupa, Meera Unnikrishnan

TL;DR
This study shows that the EsxC protein in Staphylococcus aureus affects how well daptomycin works by controlling the bacteria's cell membrane structure.
Contribution
The study reveals a novel role of EsxC in modulating membrane integrity and antibiotic sensitivity in S. aureus.
Findings
EsxC-deficient S. aureus strains are more sensitive to daptomycin and other membrane-targeting antibiotics.
The absence of EsxC alters membrane properties, including charge and fluidity, increasing daptomycin binding.
EsxC influences calcium ion interactions with the membrane, affecting antibiotic activity and infection outcomes.
Abstract
The human pathogen Staphylococcus aureus encodes a specialized type VII secretion system (T7SS), which plays an important role in bacterial virulence during infection. However, the functions of the T7SS during infection and in bacterial physiology remain unclear. Here, we demonstrate that S. aureus strains lacking a T7SS effector, EsxC (ΔesxC), were highly sensitive to the important last resort drug, daptomycin, as well as other membrane-targeting antibiotics, including gramicidin and bithionol. To understand how EsxC mediates increased antibiotic sensitivity, we investigated the ΔesxC cell envelope. Scanning electron microscopy analysis of the esxC mutant revealed a distinct cell surface morphology. ΔesxC also displayed a decrease in membrane fluidity, altered membrane protein profiles, and altered cell wall synthesis. The esxC mutant membranes demonstrated an increased negative charge…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
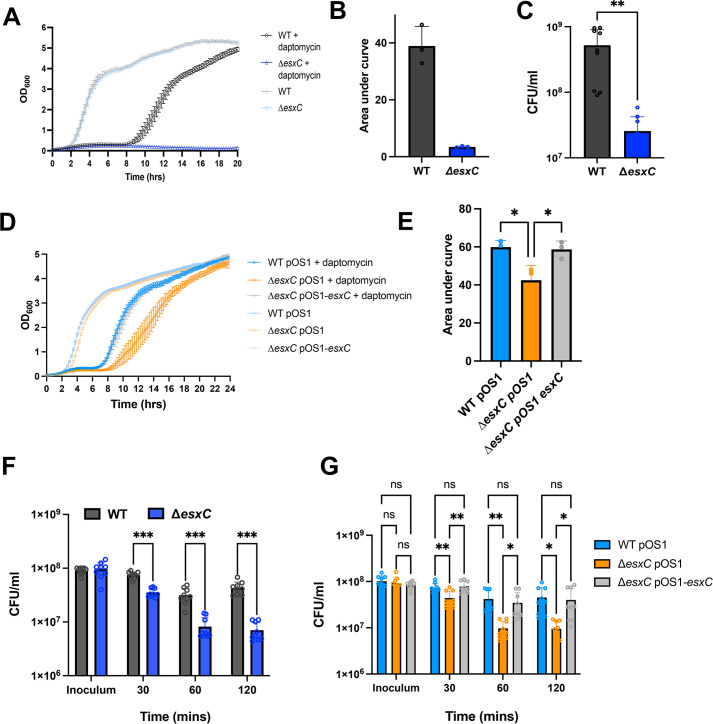 Fig 1
Fig 1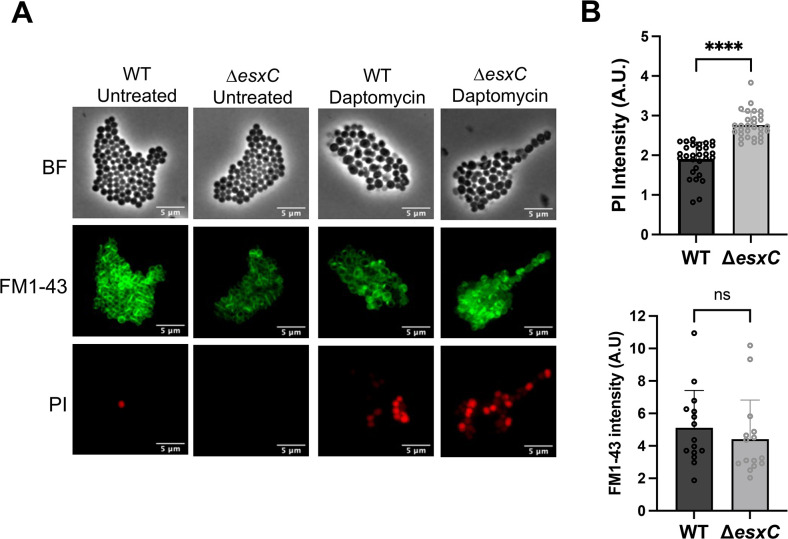 Fig 2
Fig 2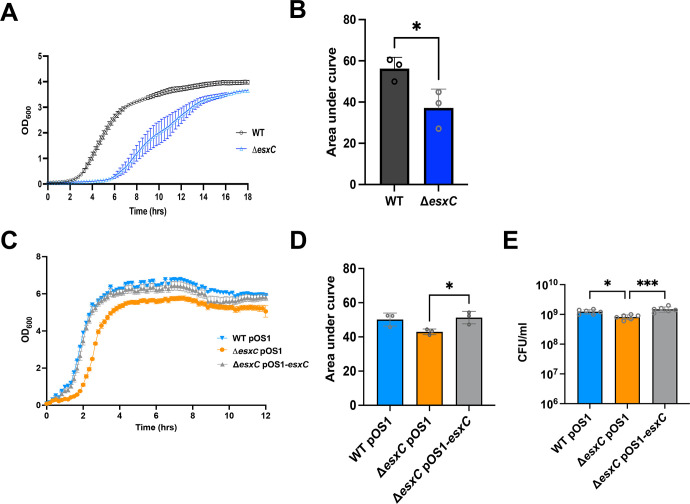 Fig 3
Fig 3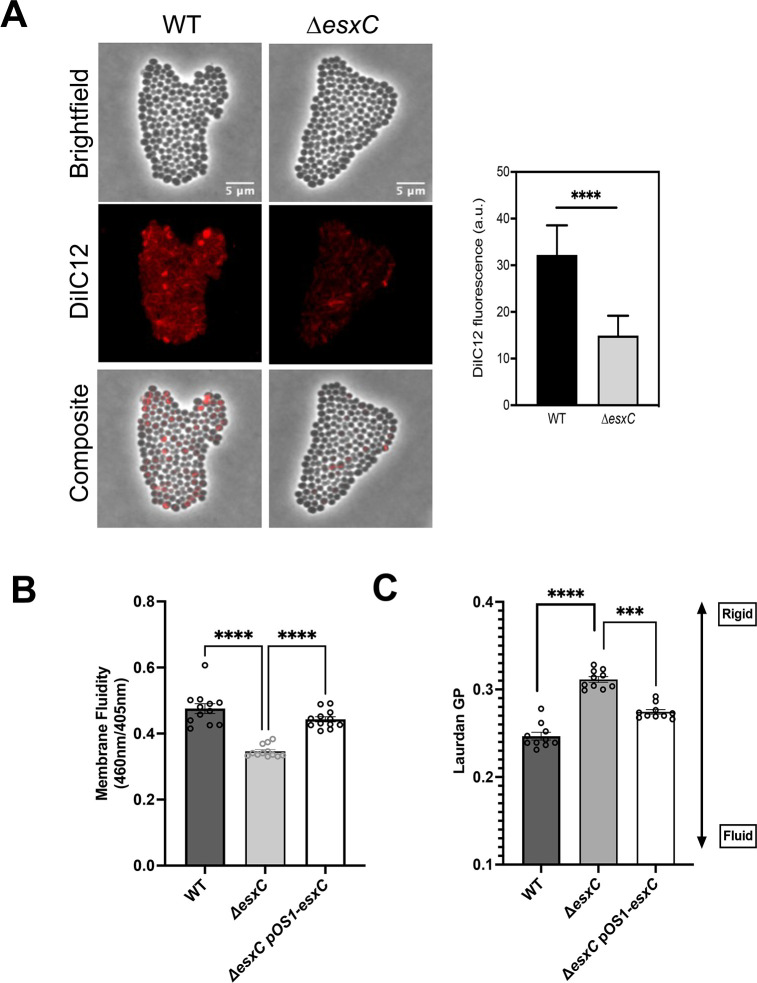 Fig 4
Fig 4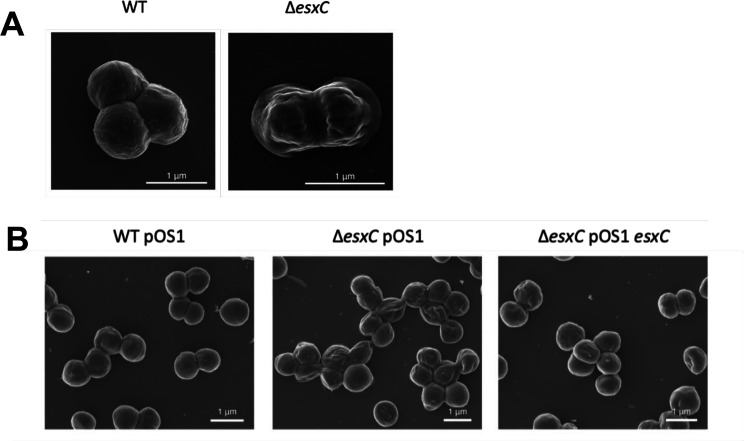 Fig 5
Fig 5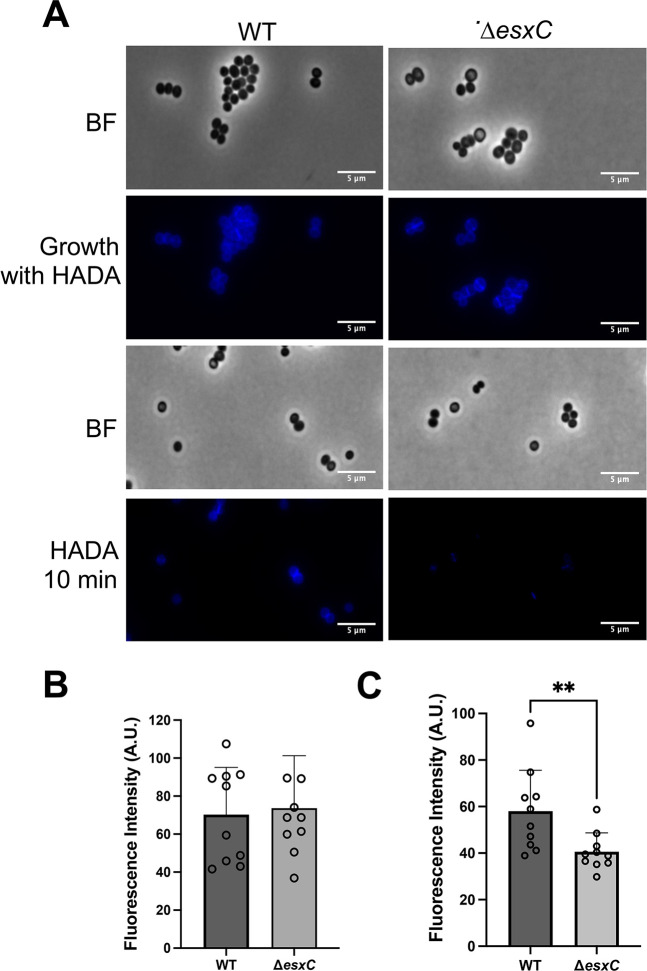 Fig 6
Fig 6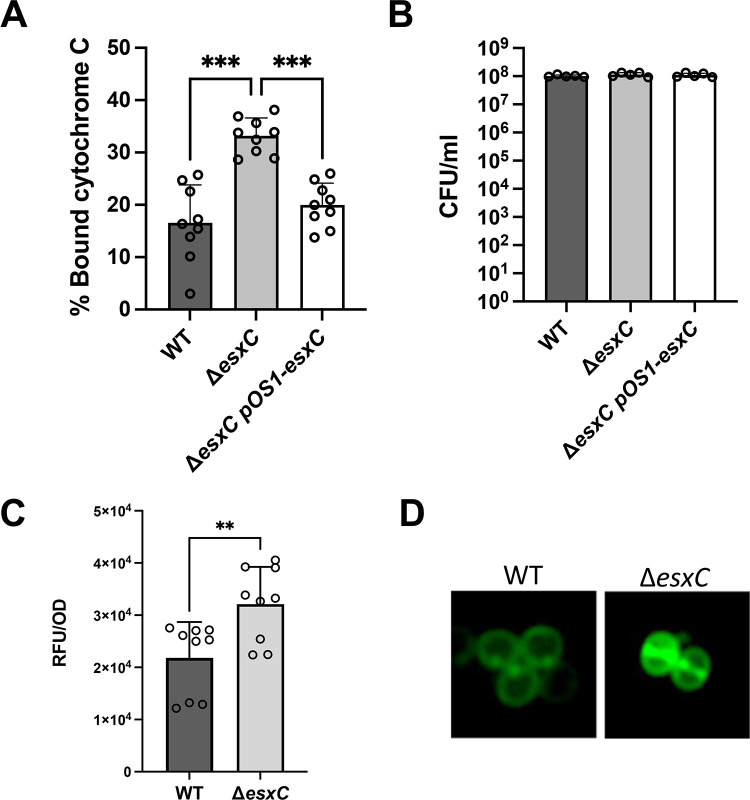 Fig 7
Fig 7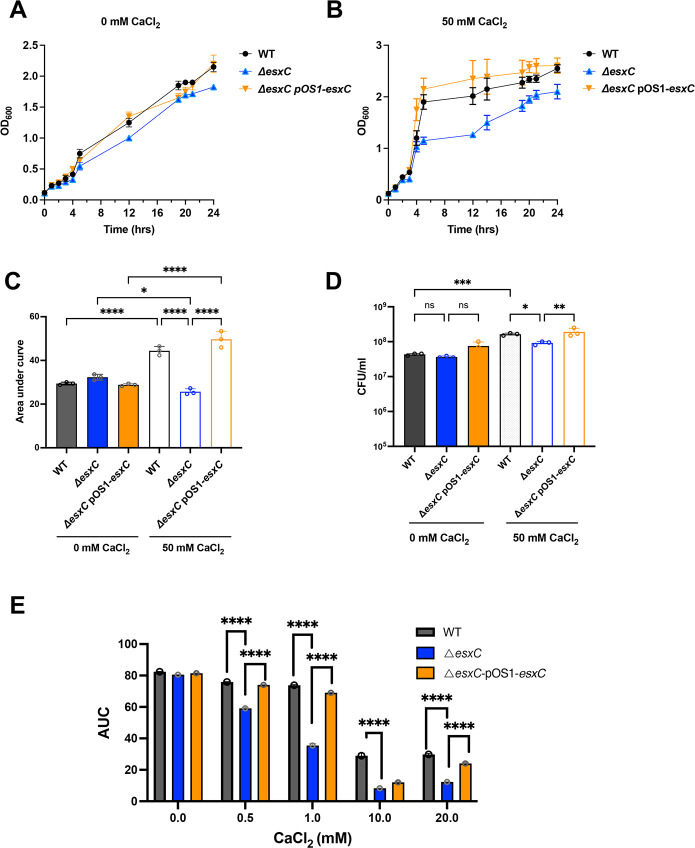 Fig 8
Fig 8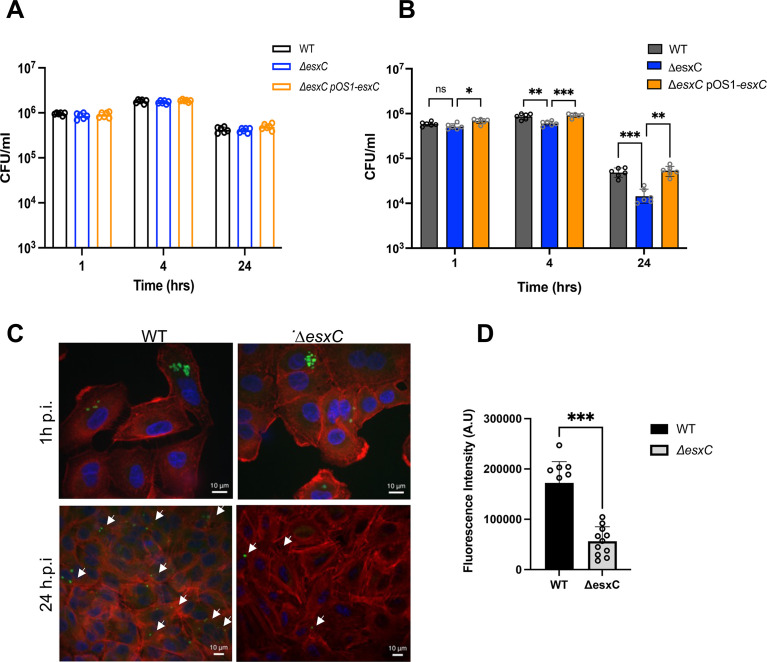 Fig 9
Fig 9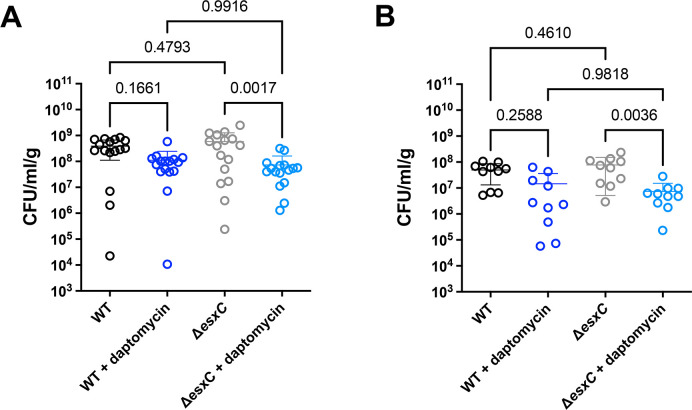 Fig 10
Fig 10| Uniprot ID | log2 fold change | Adjusted | Description |
|---|---|---|---|
| A0A0H2XJR2 | −5.46 | 6.21E-08 | Pyridine nucleotide-disulfide oxidoreductase |
| A0A0H2XJ15 | −5.34 | 3.80E-09 | TPR domain protein |
| A0A0H2XIV9 | −5.33 | 1.49E-07 | T7SS protein, EsaD |
| A0A0H2XJ90 | −4.82 | 6.92E-09 | D-isomer specific 2-hydroxyacid dehydrogenase family protein |
| A0A0H2XF36 | 3.62 | 2.14E-08 | Uncharacterized protein |
| A0A0H2XJV3 | 3.67 | 2.26E-08 | ABC transporter, ATP-binding protein |
| A0A0H2XGS2 | 3.70 | 1.71E-08 | Putative sensor histidine kinase |
| A0A0H2XG87 | 4.01 | 1.59E-08 | Bifunctional ligase/repressor BirA |
| A0A0H2XH85 | 4.14 | 1.04E-08 | Cobalamin synthesis protein/P47K family protein |
| A0A0H2XER9 | 4.38 | 9.36E-09 | LysR family regulatory protein |
| A0A0H2XEB7 | 5.25 | 2.39E-07 | Integral membrane protein |
| A0A0H2XJ96 | 5.27 | 1.04E-08 | NADH-dependent flavin oxidoreductase |
| A0A0H2XHU5 | 5.97 | 2.55E-09 | Putative membrane protein |
| Strain or plasmid | Description | Source or reference |
|---|---|---|
|
| ||
| USA300 JE2 | Plasmid-cured USA300 LAC | BEI Resources (NARSA) |
| USA300 JE2 Δ | USA300 LAC JE2 defective for EssC | ( |
| USA300 JE2 Δ | USA300 LAC JE2 defective for EsxC | ( |
| USA300 LAC | Community acquired MRSA | ( |
| USA300 LAC Δ | USA300 LAC defective for EsxA | ( |
| USA300 LAC Δ | USA300 LAC defective for EsxB | ( |
| Newman | Methicillin-sensitive | ( |
| Newman Δ | Newman defective for EsxA | ( |
| Newman Δ | Newman defective for EsxB | ( |
| RN6390 | NCTC8325 derivative, Δ | Tracy Palmer ( |
| RN6390 Δ | RN6390 defective for EssC | Tracy Palmer ( |
| RN6390 Δ | RN6390 defective for EsxC | Tracy Palmer ( |
| Plasmids | ||
| pOS1 | Empty vector for complementation | Olaf Schneewind ( |
| pOS1- | ( | |
- —Medical Research Councilhttp://dx.doi.org/10.13039/501100007155
- —Biotechnology and Biological Sciences Research Councilhttp://dx.doi.org/10.13039/501100000268
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Biochemical and Structural Characterization · Streptococcal Infections and Treatments
INTRODUCTION
Staphylococcus aureus is a highly versatile pathogen and a commensal bacterium that causes infections in both humans and animals. Staphylococcal infections can be nosocomial or community-acquired and can range from mild skin abscesses to severe life-threatening infections, including septic shock and bacteremia (1). S. aureus infections place a huge cost burden on healthcare sectors across the world, especially due to the occurrence of antibiotic-resistant strains, such as methicillin-resistant S. aureus (MRSA) and strains with varying levels of vancomycin resistance (2, 3).
S. aureus possesses an array of surface and secreted factors that help the bacterium colonize the host and establish an infection (4). One such virulence-associated factor is the type VII secretion system (T7SS), which is widely found in Gram-positive bacteria (5). The T7SSb in S. aureus and other firmicutes is distinct from the systems in mycobacteria (T7SSa), although both are characterized by small substrates containing the WXG motif, such as EsxA and EsxB (6, 7). The T7SS in S. aureus is modular in nature, with heterogeneous expression between strains (8, 9). Typically, in commonly studied S. aureus strains, including USA300, Newman, and RN6390, the T7SS locus comprises integral membrane proteins (EsaA, EssA, EssB, and EssC), cytosolic proteins (EsaB and EsaG), several secreted substrates (EsxA, EsxB, EsxC, EsxD, and EsaD), and chaperones (EsaE) (7, 10). EssC is the central transporter, which is responsible for export of substrates and co-dependent export of secreted substrates, has been reported (6, 11). While specific interactions between the different T7SS components have been described, the overall structure of the secretion apparatus is poorly defined for the S. aureus T7SS (10, 11). Assembly of a functional system is dependent on the membrane microdomain protein, flotillin A (FloA) (12).
T7SS has been reported to play a role in the modulation of intraspecies competition. A T7SS substrate, EsaD, with nuclease activity mediates killing of S. aureus strains that lack the anti-toxin EsaG (13). An LXG-domain containing protein, TspA, encoded distally to the T7SS cluster shows T7SS-dependent toxic activity, which can be neutralized by an antitoxin TsaI (14). Recent work has shown that the secretion of the nuclease toxin EsaD was facilitated by EsxD, EsxC, and EsxB, which formed a pre-secretion complex (15). Some T7 components modulate host interactions during infection; EsxA modulates host cell death, and EsaE elicits cytokine production during infection (16, 17). However, effectors of this system are yet to be defined in many S. aureus strains, and their specific functions remain elusive.
T7SS in other bacterial species has been described to support bacterial functions like DNA transfer, membrane integrity, and spore development, although for S. aureus, it is not clear if T7SS is important in staphylococcal physiology (18–20). The T7SS substrate EsxC is a small WXG-like protein, expressed and important during persistent murine S. aureus infection (21). Our recent study showed that a mutant lacking EsxC was more sensitive to unsaturated host-derived fatty acids (22). T7SS defects compromised membrane integrity and induced oxidative stress in the presence of antimicrobial fatty acids, indicating a role for these proteins in membrane homeostasis. Here, we report that the absence of EsxC causes an increased sensitivity to membrane targeting drugs like daptomycin, both during infection in vitro and in vivo. We demonstrate distinct effects on cell surface morphology, membrane fluidity, and charge, indicating a role for T7SS in controlling membrane integrity potentially through modulating calcium binding. Our data suggest that targeting the T7SS could augment activities of membrane-acting drugs like daptomycin.
RESULTS
A mutant lacking T7SS effector EsxC is more sensitive to membrane-acting drugs
The cyclic lipopeptide antibiotic, daptomycin, is a key drug used to treat resilient S. aureus infections. Daptomycin binds to the cell membrane in a calcium-dependent manner (23). As our previous studies indicated a potential impact of T7SS, in particular the effector EsxC, on membrane homeostasis (22), we investigated whether the S. aureus USA300 JE2 mutants lacking the T7SS substrate EsxC (ΔesxC) respond differently to membrane-acting drugs like daptomycin. When cultured in the presence of 5 μg/mL daptomycin, growth of the wild-type (WT) USA300 JE2 strain was delayed, but growth of USA300 JE2 ΔesxC was significantly impaired compared to the WT (Fig. 1A and B). No differences in growth rates were seen between the strains in tryptic soy broth (TSB) without antibiotic. Further, a 10-fold decrease in colony-forming unit counts for the mutant was also observed in the presence of daptomycin (Fig. 1C). Complementing ΔesxC with pOS1-esxC restored WT phenotype (Fig. 1D and E), although daptomycin did not inhibit the pOS1 empty plasmid-bearing ΔesxC to the same extent as ΔesxC without plasmid. This could be attributed to differences in growth profiles of pOS1 containing WT and mutant strains, possibly due to effects of chloramphenicol exposure prior to performing growth assays. Next, in a daptomycin killing assay where logarithmic phase bacteria were treated for 2 h with daptomycin, ΔesxC was more sensitive to daptomycin compared to the WT, with the ΔesxC pOS1-esxC strain reversing the daptomycin sensitivity (Fig. 1F and G). When we stained bacteria with propidium iodide (PI), a fluorescent intercalating agent that cannot pass through the membranes of viable cells, cytosols of daptomycin-treated ΔesxC showed a higher intensity of PI compared to the WT, as quantified by fluorescence microscopy (Fig. 2A and B). Staining with the lipid staining dye FM1-43 was of similar intensity for WT and ΔesxC (Fig. 2A and B). Thus, ΔesxC demonstrated increased membrane permeability in the presence of daptomycin.
*An esxC mutant is highly sensitive to daptomycin. (A) Growth curves of S. aureus WT USA300 JE2 and ΔesxC in TSB in the absence or presence of 5 μg/mL daptomycin and 1 mM CaCl2. Mean ± standard error of the mean (SEM) is shown, N = 3 (biological replicates). AUC was calculated for the strains grown in the presence of daptomycin (B). (C) CFU/mL was enumerated after 8 h daptomycin treatment. N = 3 (biological replicates), mean ± standard deviation (SD), ***P ≤ 0.001 using an unpaired t-test. (D) Growth curves of USA300 JE2 WTpOS1, ΔesxCpOS1, and complemented strain, ΔesxCpOS1-esxC in TSB supplemented with 5 μg/mL daptomycin and 1 mM CaCl2. Mean ± SEM shown, N = 3 (biological replicates); AUC was calculated for the strains grown in the presence of daptomycin (E), mean ± SD **P ≤ 0.05 using a one-way ANOVA with Tukey’s multiple comparison test. (F) CFU/mL of WT USA300 JE2 and ΔesxC was determined from a killing assay in the presence of 10 μg/mL daptomycin and 1 mM CaCl2 at different time points. N = 3 (biological replicates), (G) CFU/mL from killing assay using WT USA300 JE2, ΔesxC, and ΔesxC complemented strains in the absence or presence of 10 μg/mL daptomycin and 1 mM CaCl2 at different timepoints, mean ± SD, N = 3, *P ≤ 0.05, **P ≤ 0.01, **P ≤ 0.001 using a two-way ANOVA with a Tukey’s multiple comparison test for (F) and (G).
*Membrane permeability of the esxC mutant in the presence of daptomycin. (A) Representative fluorescent micrographs of S. aureus USA300 JE2 WT and ΔesxC treated with daptomycin. Cell membranes were stained by FM1-43 and imaged using FITC filter channel, PI was imaged using Texas red filter channel. (B) Images of daptomycin-treated samples in (A) were quantified using FIJI. Mean fluorescence intensity was quantified from 5–10 images from each experiment and normalized to the number of cells in the field of view. Data presented are from three independent experiments. ***P < 0.0001 using an unpaired t test with Welch’s correction. ns, non significant.
To investigate whether other T7SS effectors had similar effects, we studied strains lacking substrates EsxA and EsxB using mutants in USA300 LAC and Newman backgrounds. Growth defects were observed in USA300 LAC and Newman ΔesxA mutants compared with the respective WT strains, while ΔesxB mutants did not appear to have a growth defect (Fig. S1A through D). The Newman strain was more sensitive than USA300 JE2 to daptomycin, with growth delayed until ~14 h. Furthermore, a USA300 JE2 mutant lacking another T7SS substrate, EsxD, did not display any growth defects in the presence of daptomycin (Fig. S1E and F). Also, a USA300 JE2 strain lacking the central transporter EssC (ΔessC) responsible for transport of T7SS effectors, as expected, showed significantly impaired growth compared with the WT (Fig. S1G and H). Complementation of daptomycin sensitivity was seen for ΔesxA and ΔessC (Fig. S1K through N). Furthermore, ΔesxC and ΔessC mutants in a S. aureus RN6390 background were more susceptible than the WT to daptomycin, although bacterial growth initiated much later (~14 h) compared with the JE2 strain (~8 h) (Fig. S1I and J).
To determine if ΔesxC was sensitive to other membrane-targeting antimicrobials, gramicidin, a polypeptide antimicrobial peptide that forms ion channels in the cell membrane (24), and bithionol, an inhibitor of soluble adenylyl cyclase and its membrane-disrupting activity, were tested (25). The WT JE2 strain did not show any growth defects when treated with 2 μg/mL gramicidin; however, the ΔesxC mutant displayed slower growth (Fig. 3A and B). Complementation reversed the phenotype, but as with daptomycin, the plasmid containing the ΔesxC mutant strain showed a reduced sensitivity as compared with the ΔesxC mutant (Fig. 3C through E). A slightly slower growth was also seen for the mutant in the presence of 1 μg/mL bithionol compared with the WT (Fig. S2A through C). Finally, this effect appears to be specific to membrane-acting drugs as drugs targeting the cell wall (vancomycin, oxacillin) or DNA replication (ciprofloxacin, mitomycin) did not differentially affect the growth of the ΔesxC (or ΔessC) as compared with the WT (Fig. S2D through G).
*The esxC mutant shows higher sensitivity to other membrane-acting drugs. Growth curves of USA300 JE2 WT and ΔesxC (A) and (C) WTpOS1, ΔesxC pOS1, and ΔesxCpOS1esxC plasmid-complemented strains in the presence of 2 μg/mL gramicidin, mean ± SEM, N = 3 (biological replicates). (B and D) Area under curves (AUC) were calculated, and the mean AUC ± SD was plotted for growth curves in (A) and (C), **P = 0.001 using an ordinary one-way ANOVA. (E) CFU/mL was enumerated after 4 h gramicidin treatment. N = 3 (biological replicates), mean ± SD is shown, **P = 0.004 using a one-way ANOVA and Sidak’s multiple comparisons test.
ΔesxC displays decreased membrane fluidity and altered membrane protein profiles
Changes in membrane fluidity can affect membrane function and antibiotic sensitivity. We employed multiple assays to assess the membrane fluidity of the ΔesxC mutant. The WT strain stained with a fluorescent membrane dye DiI12C, which detects regions of increased fluidity in bacteria (26), showed a few fluid regions with uneven membrane staining, as reported previously. In contrast, the ΔesxC mutant displayed weak membrane staining (Fig. 4A), which may indicate a decrease in membrane fluidity. In an alternate membrane fluidity assay using pyrene decanoic acid, an excimer-forming lipid (27), compared with the WT, the ΔesxC mutant membranes showed a mild but statistically significant increase in rigidity, which was reversed upon complementation of esxC (Fig. 4B). The plasmid containing WT and mutant strains did not behave differently, unlike growth curve experiments in [Fig. 1 and 3](#F1 F3). Finally, membrane fluidity measurements using the fluorescent dye Laurdan (6-dodecanoyl-2-dimethylaminonaphthalene), which is incorporated into the membrane bilayer (28), revealed a lower average generalized polarization value of 0.33 for WT compared with 0.42 for ΔesxC, which was reversed in ΔesxC pOS1esxC, suggesting that the membrane of the ΔesxC mutant is more rigid in comparison to that of the WT (Fig. 4C). Overall, the data indicate that EsxC contributes to modulation of S. aureus membrane fluidity.
*EsxC affects membrane fluidity. (A) Widefield micrographs of S. aureus WT USA300 JE2 and ΔesxC after growth in TSB to OD600 of 1.0 in the presence of the lipophilic dye DiIC12. Images are representative of 4 independent experiments. The DiIC12 fluorescence of 80 bacterial clusters from different fields per strain was quantitated with ImageJ. Means ± SD are shown, N = 4 (biological replicates); ****P < 0.0001, using an unpaired t-test. (B) The membrane fluidity of S. aureus WT USA300 JE2, ΔesxC, WTpOS1, ΔesxC pOS1, and ΔesxCpOS1esxC as measured with a pyrene decanoic acid staining-based assay, mean ± SEM is shown, N = 6 (biological replicates), * indicates P < 0.05 using a one-way ANOVA with Tukey’s multiple comparisons test. (C) USA300 JE2 WT, ΔesxC, WTpOS1, ΔesxC pOS1, and ΔesxCpOS1esxC were grown to logarithmic phase in TSB at 37°C prior to staining with Laurdan. To assess membrane fluidity, Laurdan generalized polarization (GP) was calculated using the equation (I460 – I500)/ (I460 I500), where I refers to the fluorescence intensity at the indicated emission wavelength, mean ± SD is shown, N = 3 (biological replicates), **P <0.0001 using a one-way ANOVA with Tukey’s multiple comparisons test.
Previously, we reported that ΔesxC and ΔessC had distinct total protein profiles with and without treatment with linoleic acid (22). Given the changes seen in bacterial membranes in the absence of EsxC, we examined global changes in the protein content in the membrane. Membrane fractions extracted from logarithmic phase (OD_600_ = 3.0) cultures of WT and ΔesxC were analyzed by nanoLC-ESI-MS/MS. Thirteen proteins were altered in abundance [log2 (fold change) = 1.0, P < 0.05] in ΔesxC in relation to the WT (Table 1). Membrane proteins were increased in abundance, and EsaD, a T7SS nuclease, was decreased significantly in the membranes of ΔesxC. A secretome analysis from culture supernatants showed a lower abundance of substrates EsxA, EsaD, and LXG toxin TspA (A0A0H2XH53) (14) in the ΔesxC compared with WT (Table S1). Thus, the esxC mutant shows distinct changes to the overall membrane protein profiles and secretion of certain T7SS substrates appears to be co-dependent on substrate EsxC (15).
EsxC contributes to altered cell surface morphology and decreased rate of cell wall synthesis
As EsxC appeared to be involved in membrane function, we next examined the cell surface of ΔesxC using scanning electron microscopy. The cell surface of early logarithmic phase WT showed a typical smooth spherical shape; however, ΔesxC appeared deflated with dents or hollows (Fig. 5A). The WT phenotype was restored in the ΔesxC pOS1-esxC complemented strain (Fig. 5B). Interestingly, this cell surface defect was not seen in ΔesxD (Fig. S3). S. aureus cell surface alterations were seen in USA300 (LAC) and Newman strains lacking EsxA (ΔesxA) but not in ΔesxB (Fig. S3). However, RN6390 strains lacking esxC showed very little surface defects (Fig. S3). Finally, the JE2 transporter mutant ΔessC showed defects similar to ΔesxC (Fig. S3). Differences seen in surface morphologies were not due to growth rate differences, as growth rates of all the mutant strains in TSB were similar to that of their respective WT counterparts (Fig. S1). Our data suggest that the cell envelopes of mutants lacking specific T7SS components were altered, potentially due to a defect in the cell membrane and/or cell wall structure.
The esxC mutant displays a distinct surface morphology. (A) Representative SEM images of S. aureus WT USA300 JE2 and ΔesxC grown to early logarithmic phase. (B) Representative SEM images of S. aureus USA300 JE2 WT pOS1, ΔesxC pOS1, and ΔesxC pOS1-esxC grown to early logarithmic phase.
Transmission electron microscopy was used to investigate if the surface structure changes were associated with defective cell wall structure or synthesis, but no clear differences in cell wall thickness were seen between strains (Fig. S4). To study whether peptidoglycan synthesis and accumulation were affected in the T7SS mutants, the fluorescent D-amino acid, 7-hydroxycoumarincarbonylamino-D-alanine (HADA), was used (29). No differences were observed in the cells, which were previously grown to log phase and supplemented with HADA, a fluorescent D-amino acid (Fig. 6A and B). However, the addition of HADA during exponential phase growth revealed a faster accumulation of HADA in the WT compared with ΔesxC (Fig. 6A and C). This suggests that ΔesxC displayed a decreased rate of new peptidoglycan synthesis.
*EsxC impacts cell wall synthesis. (A) Representative widefield micrographs of S. aureus WT USA300 JE2 and ΔesxC either grown to logarithmic phase in the presence of 25 μM HADA or after 10-min treatment with HADA once OD600 of 1 was reached. (B) Quantification of fluorescence in cells grown to logarithmic phase with HADA. (C) Quantification of fluorescence in cells treated with HADA for 10 min. Quantification was performed on five images each from three independent experiments. *P< 0.01 and using an unpaired t-test.
ΔesxC displays an increased negative cell surface charge and increased binding of daptomycin
As we observed cell surface defects and changes to peptidoglycan synthesis, we tested if the deletion of esxC could alter other cell surface properties like surface charge. A cytochrome binding assay was employed to measure surface charge; the binding of cytochrome C, a highly positively charged protein, is directly proportional to the net negative surface charge of S. aureus cells (30). A significantly higher level of binding of cytochrome C to the cell surface of ΔesxC and ΔesxA was observed compared with the respective WT (Fig. 7A; Fig. S5), suggesting that the cell surfaces of ΔesxC and ΔesxA were more negatively charged. Much lower levels of cytochrome C bound to USA300 LAC WT and ΔesxB, suggesting these cell surfaces are less negative (Fig. S5). CFU counts from samples after cytochrome C addition showed no differences in cell numbers, and therefore, it is unlikely that bacterial cell numbers influenced cytochrome C binding (Fig. 7B; Fig. S5). These data indicate that the lack of T7SS effector EsxC impacts the surface charge of S. aureus.
*EsxC mutant membranes are more negatively charged. (A) Quantitative binding assay of cytochrome C to USA300 JE2 WT, ΔesxC, and ΔesxCpOS1-esxC. The CFU of the starting inoculum was calculated for each mutant (B). Mean ± SD shown, N = 3 (biological replicates), ***P < 0.001 using an ordinary one-way ANOVA with a Tukey’s multiple comparisons test. (C) Fluorescence measurements of daptomycin-BODIPY binding to S. aureus WT USA300 JE2 and ΔesxC. The excitation wavelength was 488 nm, and emission wavelength was 530 nm. N = 3 (biological replicates), mean ± SD. *P = 0.0067, using an unpaired t-test. (D) Representative micrographs of daptomycin-BODIPY binding to S. aureus WT and ΔesxC are shown.
S. aureus has been reported to resist daptomycin through possessing a more positive surface charge (31, 32). Based on the increased negative surface charge observed for the T7SS mutants, we hypothesized that daptomycin could bind more easily in the absence of a functional T7SS. To test this, BODIPY-tagged daptomycin was used to quantify binding via fluorometry (Fig. 7C) and fluorescence microscopy (Fig. 7D). In the presence of daptomycin, a higher level of daptomycin was observed to bind around the septum for the WT as reported previously (33), while ΔesxC showed a significantly higher fluorescence. Thus, our data suggest that reduced electrostatic repulsion of daptomycin may contribute to the increased daptomycin binding in ΔesxC.
Calcium affects the growth of ΔesxC and sensitivity to daptomycin
Calcium is an important component in membranes and affects both structure and charge of bacterial membranes (34). Calcium is also required for the activity of daptomycin and can enhance killing by gramicidin (35, 36). We hypothesized that EsxC may control membrane integrity through modulating the membrane binding of calcium. To test this, EsxC mutants were cultured in minimal medium with or without calcium chloride (50 mM). While both WT and mutant strains showed slow growth in minimal medium in the absence of calcium, in the presence of 50 mM calcium chloride, the mutant grew much slower than the WT and the esxC-complemented strains, with lower CFUs recovered after 4 h growth (Fig. 8A through D). We then examined if the daptomycin toxicity was dependent on the concentration of calcium available. WT, ΔesxC, and the complemented strains were cultured in the presence of 5 μg/mL daptomycin and increasing calcium chloride concentrations (0 to 20 mM). While daptomycin did not have any effect in the absence of calcium as expected, a dose-dependent decrease in growth of the WT strain was observed in CaCl_2_, with the WT showing slower growth at 10 and 20 mM CaCl_2_ compared with 1 mM (Fig. 8E; Fig. S6). The mutant was more sensitive to daptomycin at all calcium concentrations compared with the WT and complemented strains, with increased growth inhibition at higher CaCl_2_ concentrations.
*Calcium affects growth of esxC mutant and daptomycin sensitivity growth curves of S. aureus WT USA300 JE2, ΔesxC, and ΔesxC pOS1-esxC in synthetic minimal medium in the absence (A) or presence (B) of 50 mM CaCl2, mean ± SEM is shown. (C) AUC was calculated from (A and B) and the mean AUC ± SD was plotted. Mean ± SD, *P = 0.011, ***P < 0.001, using ordinary one-way ANOVA with Tukey’s multiple comparisons test, (D) CFU was enumerated after 5 h growth in absence or presence of 50 mM CaCl2., mean ± SD, *P = 0.03, **P = 0.001, ***P = 0.0004, ns - non-significant using ordinary one-way ANOVA, with a multiple Tukey’s comparisons test. (E) S. aureus WT USA300 JE2, ΔesxC, and ΔesxC pOS1-esxC were grown in TSB supplemented with increasing calcium chloride concentrations (0 to 20 mM) in the presence of daptomycin (5 µg/mL). AUCs calculated from growth curves shown in Fig. S6 are shown. mean ± SD, ***P = 0.0001, ***P < 0.0001, using two-way ANOVA with a Tukey’s multiple comparison test.
Hence, calcium binding to membranes and/or downstream signaling may be impacted in the ΔesxC, affecting its ability to grow in the presence of calcium and its sensitivity to membrane-targeting drugs like daptomycin.
Decreased intracellular survival of ΔesxC in the presence of daptomycin
Daptomycin has been shown to kill intracellular S. aureus less effectively (37). To determine if deletion of esxC from S. aureus would enhance intracellular killing activity of daptomycin, A549 human lung epithelial cells (16) treated with a high concentration of daptomycin (20 μg/mL) or untreated were infected with S. aureus. There were no significant differences in bacterial internalization or in intracellular bacterial survival between WT and ΔesxC in untreated epithelial cells (Fig. 9A). In the daptomycin-treated cells, while there were no differences between numbers of internalized WT and ΔesxC 1 h post infection (p.i.), a significant decrease was observed in ΔesxC survival within cells treated with daptomycin in comparison to WT, 4 h, and 24 h p.i. (Fig. 9B). Notably, there was a significant number of intracellular WT S. aureus surviving after treatment with daptomycin. Confocal microscopy of infected cells also showed more S. aureus WT at 24 h p.i. compared with ΔesxC in the presence of daptomycin (Fig. 9C and D). WT and mutant strains had similar growth rates in DMEM-10 (Fig. S7A), with a growth defect in the presence of daptomycin as expected (Fig. S7B). Therefore, our data suggest that daptomycin is more effective in killing intracellular S. aureus in the absence of T7SS.
*Presence of EsxC affects intracellular bacterial killing by daptomycin. CFU counts at various time points during epithelial cell infection assay with WT USA300 JE2 and ΔesxC after 1, 4, and 24 h infection in the absence (A) or in the presence of 20 μg/mL daptomycin (B), N = 3 (biological replicates), mean ± SD is shown, *P = 0.01, **P = 0.004, ***P = 0.002, one way ANOVA with Tukey’s multiple comparison tests. (C) Representative confocal micrographs of cells after 1 h or 24 h post-infection. DAPI (blue), F-actin (red), and S. aureus (green, white arrows). (D) Confocal image quantification from five images each from three independent experiments after 24 h. Mean ± SD of intensity is shown, **P < 0.001 using an unpaired t-test.
S. aureus lacking EsxC is sensitive to daptomycin in a skin infection model
As T7SS proteins have been reported to be important in various murine infection models (6, 8, 38), we next investigated effects of daptomycin on S. aureus infection and the impact of EsxC on daptomycin killing in vivo employing a localized murine skin abscess model. BALB/c mice were infected with S. aureus WT and ΔesxC intradermally, followed by treatment with 10 mg/kg daptomycin as described in Materials and Methods. In the non-treated groups, bacterial counts from the site of infection (lesions) showed no significant difference in bacterial numbers between WT- and ΔesxC- infected mice after 24 or 72 h of infection. There were also no differences in bacterial survival between WT- and ΔesxC-infected mice treated with daptomycin. A significant decrease was, however, seen in the numbers of ΔesxC present in the skin of daptomycin-treated mice compared to the untreated group 24 and 72 h p. i., while this was not the case for the WT-infected group. (Fig. 10A and B). No significant differences were observed between the areas of lesions measured for WT or ΔesxC after 24 or 72 h infections and in daptomycin-treated mice (Fig. S8). Although the dose of daptomycin used in the timeframe of treatment was not sufficient to treat WT S. aureus infection, these data suggest that the strain lacking EsxC was more susceptible to daptomycin in vivo compared with the WT.
Effect of daptomycin on esxC mutants in vivo. Mice were infected in the skin with WT USA300 and ΔesxC mutant, followed by treatment with 10 mg/kg daptomycin. (A) CFU/g skin tissue was counted 24 h p.i. (n = 8, two lesions/animal). Graphs show mean ± SD, and a one-way ANOVA with a Tukey’s multiple comparisons test was used to determine significance. P-values are indicated in the graph. (B) CFU/g was calculated 72 h p.i. (n = 5, two lesions/animal), mean ± SD shown, one-way ANOVA with Tukey’s multiple comparisons test; P-values are indicated in the graph.
DISCUSSION
The S. aureus T7SS has been strongly associated with staphylococcal virulence and the ability of this pathogen to persist during infection (7). Its importance for bacterial competition between S. aureus strains has been recently highlighted (13). However, the role of this system in staphylococcal physiology has been poorly defined. Here, we report that EsxC, a T7SS effector, is important for maintaining cell membrane homeostasis through modulating membrane properties including fluidity, accompanied by changes in membrane protein localization, rate of cell wall synthesis, and surface charge. Our data indicate that EsxC may mediate these membrane effects through modulating calcium binding to the membrane. Consequently, the lack of EsxC impacts activity of membrane-acting drugs including daptomycin, a key drug used to treat resilient staphylococcal infections, with modulation of the activity of daptomycin both intracellularly as well as during infection in vivo. Although we observe T7SS effector specificity in daptomycin sensitivity and membrane defects, as EsxC secretion is co-dependent on other T7SS effectors, EsxC may mediate these effects in association with other T7SS proteins.
Several studies in mycobacterial species have linked the T7SS to cell surface integrity in different ways (20, 39–42). The ESX-1 system has been previously reported to impact cell membrane permeability in M. tuberculosis, although this effect appeared to be strain specific (39). However, ESX-5, which is considered essential for in vitro growth, has been linked to mycobacterial capsular, cell wall, and outer membrane permeability (20, 40, 43) and proposed to mediate transport of cell envelope proteins required for uptake of essential nutrients to the outer membrane. In S. aureus, we previously reported that the T7SS components EsxC and EssC were associated with the maintenance of membrane integrity in response to antimicrobial host fatty acids (22). We and others have previously reported that some secreted T7 effectors like EsxC and EsxA are associated with the staphylococcal cell membrane and/or cell wall (8, 22, 44). T7SS has also been previously linked to membrane fluidity; higher levels of T7SS transcription were associated with a decrease in membrane fluidity, caused by the fatty acid kinase complex, which incorporates host fatty acids into the membrane, inducing a change in membrane dynamics (27).
It is important to consider that T7SS substrates are secreted in a co-dependent manner, so EsxC-mediated effects may be attributed to another T7SS substrate like EsxA and LXG toxins like EsaD (11, 13). EsxC was reported to be part of the EsaD secretion complex, along with EsxD and EsxB, with EsxD required for EsaD secretion (15). However, mutants lacking EsxB and EsxD did not show similar phenotypes to EsxC. EsxD and EsxB did not appear to be associated with surface morphology defects by SEM, surface charge, and daptomycin sensitivity, indicating that these substrates did not play a role in the membrane homeostasis. Hence, at least in USA300, EsaD may not be involved, and EsxC likely has distinct effects. However, EsaD was depleted in esxC mutant membranes and supernatants. We cannot rule out that EsaD contributes to the defects seen. On the other hand, common phenotypes were seen for strains, which lacked EsxC or EsxA. Previous reports have suggested that EsxC and EsxA can interact (11), and decreased EsxA was seen in the ΔesxC secretome, indicating co-dependent secretion of EsxA and EsxC as expected (8, 11, 22). Therefore, interactions between EsxC and EsxA may be important in modulating membrane integrity. While esxA is conserved across all S. aureus strains, esxC is only found in the essC1 locus (8, 9). It is, however, likely that all S. aureus strains encode a protein orthologous to EsxC as proteins in module 2, with variants essC2-4, have not yet been characterized and could function like EsxC. Finally, in the RN6390 strain background, esxC and essC did not show similar differences to the USA300 strains by SEM, which may be due to differences in global regulatory circuits that may control cell wall synthesis (Agr, Sar) (45–47).
To explain the EsxC-associated membrane defects, multiple possibilities were considered. First, EsxC could affect other membrane-associated proteins, impacting lipid packing and fluidity of the membrane; EsxC could be a chaperone for membrane proteins with a role in targeting them to the membrane. A recent bacterial two-hybrid library screening we performed, however, was not successful in identifying any direct bacterial interactors, although such interactions may be transient. Second, it is possible that EsxC may impact the state of the membrane by binding other molecules like metal ions required for maintaining stability of the membrane (48). Our data indicate that the T7SS substrates may be involved in modulating metal binding to the membrane.
Calcium and other metal ions like magnesium are known to impact structure and function of bacterial membranes (34, 49, 50). Calcium maintains bacterial outer membrane structure through binding to bacterial lipopolysaccharide and also influences bacterial membrane fluidity (51, 52). Calcium can also have antibacterial effects; higher concentrations of calcium can disrupt membrane integrity (50, 53, 54). Calcium affects the growth of S. aureus lacking EsxC, indicating that the protein may modulate calcium binding to the membrane. EsxC may directly or with other T7SS effectors or membrane proteins, bind to calcium, and/or be involved in transporting or modulating calcium binding to the cell envelope. The molecular mechanisms underlying calcium interactions of EsxC/ other T7SS substrates need further investigation.
Daptomycin is thought to bind to the cell membrane before forming a complex with cell wall biosynthesis machinery, preventing cell wall synthesis, before dispersing throughout the bacterial membrane causing depolarization (55). Daptomycin, although not a cationic antimicrobial peptide, forms a highly positively charged complex with calcium to become active (55). Increased daptomycin binding to ΔesxC compared with the WT suggests that the decreased positive surface charge of ΔesxC allows easier daptomycin binding to the cell membrane. In agreement with this, T7SS substrates may influence the free calcium available for activating daptomycin. Daptomycin also affects bacterial membrane fluidity through perturbing membrane microdomains (56), and clinical daptomycin-resistant isolates have generally been associated with increased fluidity (57, 58). Thus, EsxC could further contribute to daptomycin resistance by altering membrane fluidity and charge. Daptomycin resistance in S. aureus has been mainly associated with mutations in genes encoding for multipeptide resistance factor (MprF), a lipid biosynthetic enzyme, and the two-component system, vancomycin-resistance associated sensor/regulator (VraSR) (59–61). Proteomic analysis of mutant whole lysates did not show any modulation of proteins implicated in daptomycin resistance, and genome sequencing did not reveal any changes in the ΔesxC genome (22).
In contrast to daptomycin, bithionol and gramicidin are uncharged (24, 25); therefore, surface charge and decreased electrostatic repulsion of antibiotics is not solely responsible for increased ΔesxC sensitivity, although calcium is known to increase gramicidin activity (35). As all three antimicrobials have different mechanisms of action once bound to the membrane, before causing membrane depolarization, the subsequent steps after membrane binding appear to be important for efficacy. Therefore, daptomycin treatment, which impacts both the cell membrane and the cell wall, may cause a larger effect in the esxC mutant.
While daptomycin kills ΔesxC in vivo, at the dose and with the strains of S. aureus used, there was no impact of the EsxC in bacterial survival during acute skin infection. T7SS proteins have been previously reported to be important for bacterial virulence using invasive kidney abscess, nasal colonization and skin infection models, although studies have used different S. aureus strains (6, 8, 38). The lack of decrease in the lesion size of the T7SS mutant-infected mice treated with daptomycin, unlike the trend towards decreased lesion size observed for the WT (Fig. S7), could indicate that the increased bacterial killing may cause an altered local immune response. A recent study reported that in a S. aureus strain WU1, the T7SS was important only for invasive infection and not localized nasal colonization (62).
Currently, levels of resistance to daptomycin are low; however, with increasing resistance in other last-line antibiotics, such as vancomycin, the use of daptomycin will increase going forward, likely leading to the occurrence of more resistant isolates (63). Therefore, targeting new proteins that can enhance existing treatments could be a promising approach. Our data indicate that inhibitors of T7SS proteins or of T7SS secretion may have good potential in combination drug therapies.
MATERIALS AND METHODS
Bacterial strains
S. aureus strains used in this study are included in Table 2. Strains were cultured aerobically in tryptic soy broth (TSB) (Sigma-Aldrich) at 37°C. Overnight cultures were subcultured into fresh TSB and grown to a density of optical density (OD_600_) of 1, unless stated otherwise. For complemented strains, TSB was supplemented with 10 μg/mL chloramphenicol.
Growth curves
Overnight bacterial cultures were diluted to an OD_600_ of 0.1 in TSB or TSB supplemented with antimicrobials where specified. Bacteria were grown in a 96-well plate with shaking, and the OD_600_ was measured every 15 min with a FLUOstar OMEGA plate reader (BMG Labtech, UK). At appropriate time points, 100 μL of the samples was taken for CFU determination.
For growth curves in defined medium, overnight cultures were subcultured into minimal defined medium, prepared as described by Machado and colleagues (66), at an initial OD_600_ of 0.15, and supplemented with varying concentrations of CaCl_2_ where stated. Bacterial growth was monitored manually by measuring OD_600_ at different time points using a spectrophotometer (Biochrom) due to issues with increased bacterial clumping in this medium on microtiter plates. Then, 100 μL of the samples was also taken for CFU determination at different time points.
Laurdan staining
Membrane fluidity was measured using the fluorescent dye Laurdan (Sigma) and was based on protocols described by Wenzel and colleagues (67). Then, 1 mL aliquots of logarithmic phase S. aureus were washed in PBS. The samples were incubated with a final concentration of 100 μM Laurdan for 5 min at 37°C in the dark, washed four times in pre-warmed PBS, following which 200 μL was transferred to pre-warmed black-walled 96-well plates. Membrane fluidity was determined by using an excitation wavelength of 350 nm and measuring fluorescence at 460 and 500 nm emission wavelengths on a BioTek Cytation 5 Cell Imaging Multimode Reader (Agilent). The generalized polarization (GP) value was calculated using the following calculation, GP = (I_460_-I_500_) / (I_460_ + I_500_).
DiIC12 staining
Overnight bacterial cultures were diluted to an OD_600_ of 0.15 in TSB with 1 μg/mL 1,1′-didodecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiIC12) (Invitrogen). Cultures were grown to an OD_600_ of 1.0, centrifuged, and washed twice with fresh TSB. Samples were imaged on agarose pads using a Leica DMi8 widefield microscope (Leica Microsystems, UK).
Cytochrome C binding assay
Overnight bacterial cultures were pelleted and washed with 20 mM MOPS buffer, pH 7. The cells were resuspended in MOPS buffer at an OD_600_ of 1 and incubated with 50 μg/mL cytochrome C at room temperature for 15 min. The cells were pelleted and the remaining cytochrome C in the supernatant was measured spectrophotometrically at OD_410_ using a FLUOstar OMEGA plate reader (BMG Labtech, UK).
Invasion and survival assays
Infection assays were carried out in 24-well plates (Falcon), or RS-treated glass chamber slides (154526, Nunc, Lab Tek II). Overnight S. aureus cultures were diluted to an OD_600_ of 0.15 and grown to an OD_600_ of 1, resuspended in DMEM to 2.5 × 10^6^ CFU/mL. A549 epithelial cells were seeded at a density of 2.5 ×10^5^ 1day before being infected with S. aureus at a multiplicity of infection (MOI) of 10 for 1 h at 37°C with 5% CO_2_. To kill any extracellular bacteria, cells were washed with DMEM containing 20 μg/mL lysostaphin (Sigma-Aldrich) and 50 μg/mL gentamycin (Melford) and incubated for 30 min. Cells were washed twice with PBS and lysed with 1 mL cold sterile water before plating for CFU determination. To investigate the course of infection after extracellular bacterial killing, DMEM with 1 mM CaCl_2_ only, or DMEM with 1 mM CaCl_2_ and 10 μg/mL daptomycin were added and incubated until the desired timepoint.
Murine skin abscess model
All animal procedures were carried out as per protocols in the Project Licence PCEC27E7D. All local ethical and home office approvals were obtained for animal experimentation. For the murine skin abscess model, female 7/8-week-old BALB/c mice (Charles River Laboratories, UK) flanks were shaved before intradermal injection with 50 μL of bacterial suspension on each flank, resulting in a final bacterial count of 1–2 × 10^6^ per mouse. Overnight S. aureus cultures were subcultured, grown to OD_600_ of 1, centrifuged and washed twice with PBS, and diluted in PBS to achieve a final CFU/mL of 1–2 × 10^7^ for infection. Two hours after infection, mice were injected intravenously via the tail vein with the dose of 10 mg/kg daptomycin or PBS. This was repeated once every 24 h. During the experiment, mice were weighed daily, and any abscess formation was measured and recorded. After 24 or 72 h, mice were euthanized by CO_2_ inhalation, and skin at the infection site was incised. The skin samples were weighed and homogenized in PBS using a FastPrep (MP Biomedicals) at 4 m/s for 60 s for a total of 6 cycles. Homogenates were diluted in PBS, and CFU/mL was determined. CFU counts were normalized to lesion weight.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. 2015. Staphylococcus aureus Infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi:10.1128/CMR.00134-1426016486 PMC 4451395 · doi ↗ · pubmed ↗
- 2Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi:10.1038/nrmicro 220019680247 PMC 2871281 · doi ↗ · pubmed ↗
- 3Lee BY, Singh A, David MZ, Bartsch SM, Slayton RB, Huang SS, Zimmer SM, Potter MA, Macal CM, Lauderdale DS, Miller LG, Daum RS. 2013. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin Microbiol Infect 19:528–536. doi:10.1111/j.1469-0691.2012.03914.x 22712729 PMC 3463640 · doi ↗ · pubmed ↗
- 4Cheung GYC, Bae JS, Otto M. 2021. Pathogenicity and virulence of Staphylococcus aureus Virulence 12:547–569. doi:10.1080/21505594.2021.187868833522395 PMC 7872022 · doi ↗ · pubmed ↗
- 5Bottai D, Groschel MI, Brosch R. 2016. Type VII secretion systems in gram-positive bacteria. Curr Top Microbiol Immunol. doi:10.1007/82_2015_501526847354 · doi ↗ · pubmed ↗
- 6Burts ML, Williams WA, De Bord K, Missiakas DM. 2005. Esx A and Esx B are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci USA 102:1169–1174. doi:10.1073/pnas.040562010215657139 PMC 545836 · doi ↗ · pubmed ↗
- 7Unnikrishnan M, Constantinidou C, Palmer T, Pallen MJ. 2017. The enigmatic Esx proteins: looking beyond mycobacteria. Trends Microbiol 25:192–204. doi:10.1016/j.tim.2016.11.00427894646 · doi ↗ · pubmed ↗
- 8Kneuper H, Cao ZP, Twomey KB, Zoltner M, Jäger F, Cargill JS, Chalmers J, van der Kooi-Pol MM, van Dijl JM, Ryan RP, Hunter WN, Palmer T. 2014. Heterogeneity in ess transcriptional organization and variable contribution of the Ess/type VII protein secretion system to virulence across closely related Staphylocccus aureus strains. Mol Microbiol 93:928–943. doi:10.1111/mmi.1270725040609 PMC 4285178 · doi ↗ · pubmed ↗