Exploration of inactive metabolic pathways in Antarctic Pseudogymnoascus australis through elicitation: a genomic and metabolomic approach to investigate its biotechnological potential
Karla Leal, Juan Machuca, David Madariaga, María José Contreras, Leticia Barrientos, Kattia Nuñez-Montero, Estefanía Chavarría, Pablo Bruna, Isabel Iturrieta-González

TL;DR
This study explores the hidden chemical potential of an Antarctic fungus using genomic and metabolomic methods, revealing new compounds and pathways.
Contribution
The first annotated genome of Pseudogymnoascus australis and the activation of silent metabolic pathways through elicitation in psychrophilic fungi.
Findings
44 biosynthetic gene clusters were identified, showing diverse secondary metabolite production potential.
Elicitation with LPS in YES medium induced the highest metabolic diversity, including cyclodipeptides and betaine lipids.
The study demonstrates the effectiveness of combining genomics and metabolomics to unlock fungal chemical potential.
Abstract
This study presents the first annotated genome of the Antarctic fungus Pseudogymnoascus australis UA-032-E, revealing significant biosynthetic potential with 44 predicted biosynthetic gene clusters (BGCs) identified through antiSMASH analysis. These BGCs include nonribosomal peptide synthetases (NRPS), type I polyketide synthases (PKS), and hybrid systems, indicating a diverse capacity for secondary metabolite production. BiG-SCAPE analysis grouped these clusters into 41 gene cluster families, with most being singletons, demonstrating low genetic redundancy and high structural diversity. To activate silent pathways, we employed elicitors (NPS and LPS) across multiple culture media, successfully inducing previously undetected metabolic activity. Using an integrated LC–QTOF–MS/MS approach combined with the GNPS and SIRIUS platforms, a total of 75 features were detected, including…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
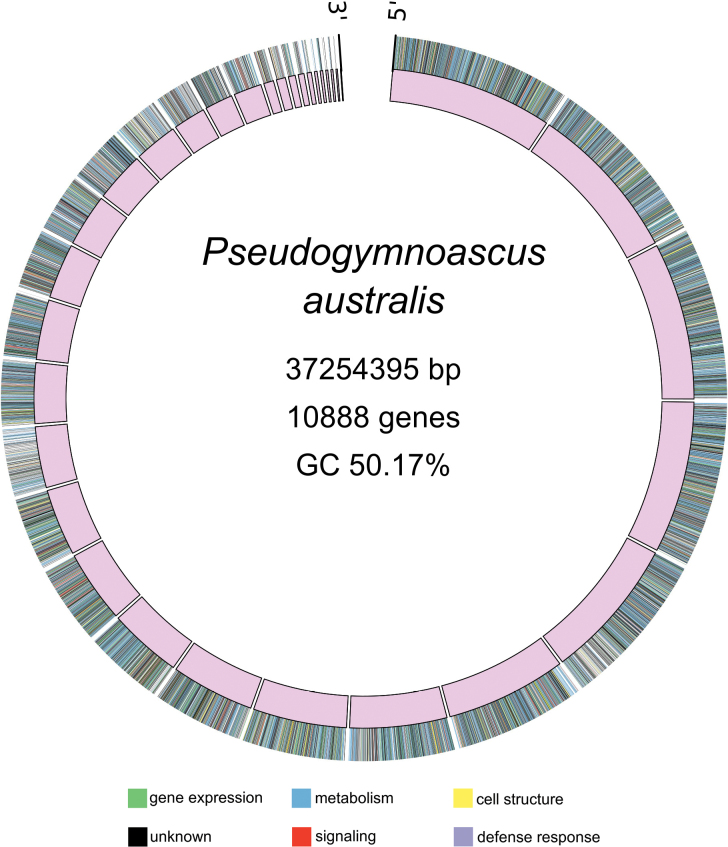 Figure 1
Figure 1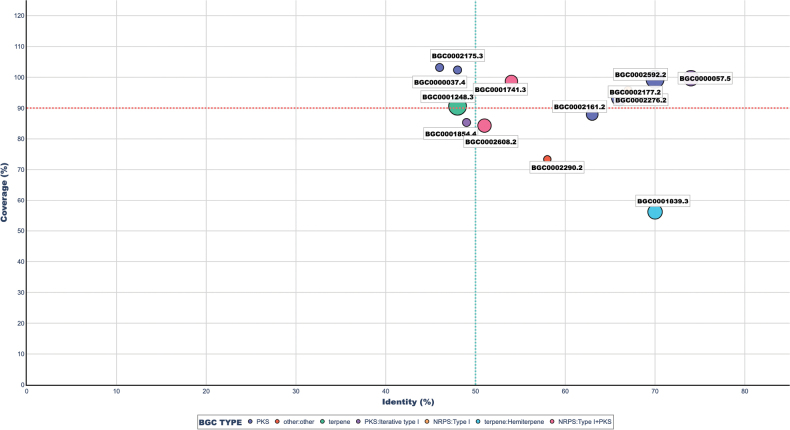 Figure 2
Figure 2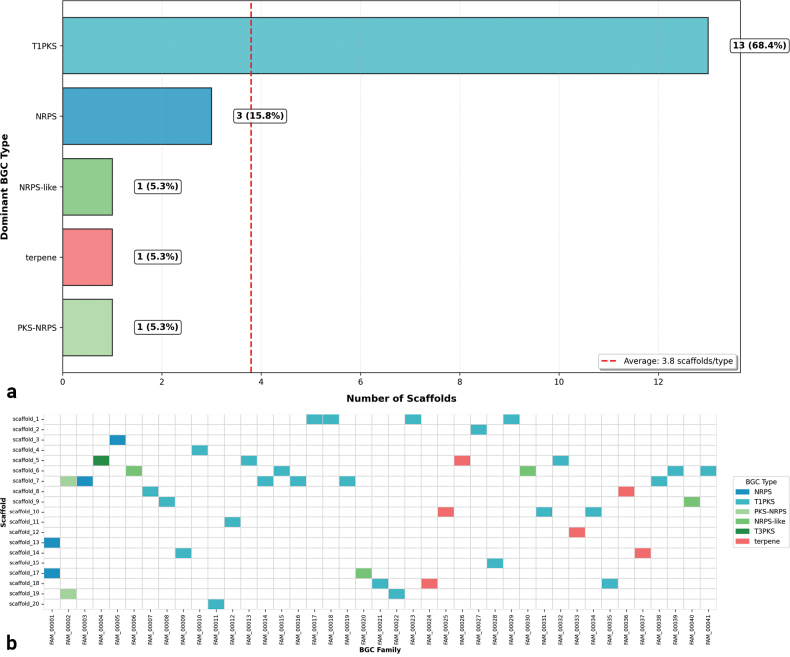 Figure 3
Figure 3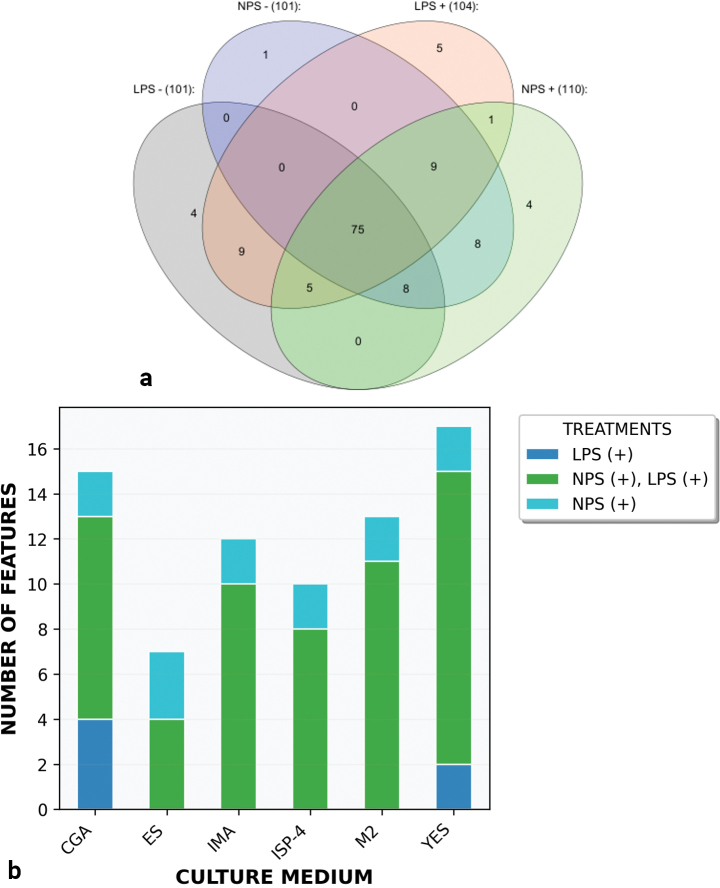 Figure 4
Figure 4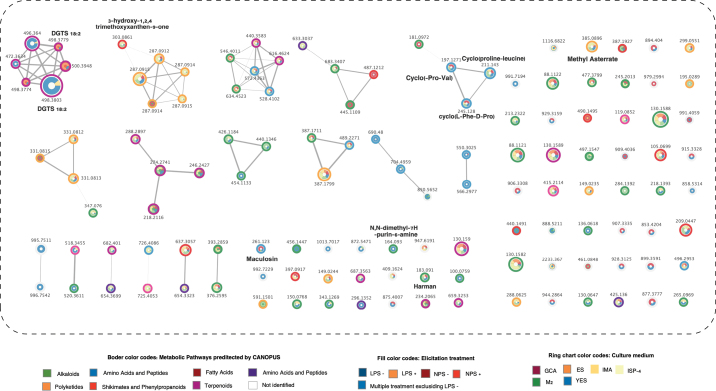 Figure 5
Figure 5| Medium | Components (per litre) |
|---|---|
| M2 | Mannitol (40 g), Maltose (40 g), Yeast extract (10 g), K2HPO4 (2 g), MgSO4·7H2O (0.5 g), FeSO4·7H2O (0.01 g) |
| IMA | Yeast extract (4 g), Malt extract (10 g), Glucose (4 g), Mannitol (40 g) |
| YES | Sucrose (150 g), Yeast extract (20 g), MgSO4·7H2O (0.5 g), ZnSO4·7H2O (0.01 g), CuSO4·5H2O (0.005 g) |
| ISP-4 | Starch (10 g), CaCO3 (2 g), (NH4)2SO4 (2 g), K2HPO4 (1 g), MgSO4·7H2O (1 g), NaCl (1 g), FeSO4·7H2O (1 mg), MnCl2·7H2O (1 mg), ZnSO4·7H2O (1 mg) |
| CGA | Glycerol (30 g), Peptone (2 g), K2HPO4 (1 g), NaCl (1 g), MgSO4·7H2O (0.5 g), Trace solution (5 mL): CaCl2·2H2O (3 g/L), MnSO4 (0.2 g/L), ZnCl2 (0.1 g/L), CuSO4·5H2O (0.025 g/L), Na2B4O7·10H2O (0.02 g/L), CoCl2 (0.004 g/L), (NH4)6Mo7O24·4H2O (0.01 g/L) |
| MIBiG Clusters | Source Compounds | Type | Total Score | Best Identity | Best Coverage | E-value |
|---|---|---|---|---|---|---|
| BGC0000037.4 | cichorine | PKS | 2122.00 | 46% | 103.18% | 0.0 |
| BGC0002290.2 | biscognienyne B / eutypinic acid | other:other | 320.00 | 58% | 73.37% | 2.28e-43 |
| BGC0001248.3 | clavaric acid | terpene | 700.00 | 48% | 90.60% | 2.61e-245 |
| BGC0000057.5 | F9775A/F9775B/orsellinic acid | PKS:Iterative type I | 4033.00 | 74% | 99.70% | 2.42e-184 |
| BGC0002276.2 | choline | 1701.00 | 67% | 95.81% | 0.0 | |
| BGC0001854.4 | tricholignan A | PKS:Iterative type I | 3623.00 | 49% | 85.32% | 0.0 |
| BGC0001839.3 | squalestatin S1 | terpene:Hemiterpene | 957.00 | 70% | 56.16% | 1.62e-162 |
| BGC0001741.3 | phyllostictine A/B | 3639.00 | 54% | 98.66% | 2.74e-120 | |
| BGC0002177.2 | ustilaginoidin N/O/M/A | PKS | 4095.00 | 66% | 93.23% | 6.55e-178 |
| BGC0002161.2 | scytalone/T3HN | PKS | 2821.00 | 63% | 87.94% | 1.95e-107 |
| BGC0002592.2 | geodin | PKS | 5323.0, 2596.0 | 70% | 99.39% | 8.42e-251 |
| BGC0002175.3 | YWA1 | PKS | 1979.00 | 48% | 102.37% | 0.0 |
| BGC0002608.2 | ochratoxin A | 412.00 | 51% | 84.31% | 1.06e-139 |
| Feature ID | Medium | Treatment | GNPS Annotation | Precursor Mass |
|---|---|---|---|---|
| 1 | YES, ES | NPS (+) | DGTS 18:2 | 498.3774 |
| 2 | YES, ES | NPS (+) | DGTS 18:2 | 498.3779 |
| 3 | YES, IMA | DGTS 18:2 | 498.3803 | |
| 4 | YES, M2, CGA, IMA | Harman | 183.0910 | |
| 5 | YES | N,N-dimethyl-7H-purin-6-amine | 164.0930 | |
| 6 | YES, M2, CGA, IMA | cyclo(L-Phe-D-Pro) | 245.1280 | |
| 7 | YES, M2, CGA, IMA | cyclo(L-Val-L-Pro) | 197.1271 |
| Feature ID | Medium | Treatment | Sirius Name | Precursor Mass |
|---|---|---|---|---|
| 1 | YES,M2,CGA,IMA | Maculosin | 261.123 | |
| 2 | YES,M2,CGA,IMA,ISP-4 | Methyl Asterrate | 385.0896 | |
| 3 | YES,M2,IMA,ISP-4 | 3-hydroxy-1,2,4-trimethoxyxanthen-9-one | 303.0861 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial Natural Products and Biosynthesis · Plant biochemistry and biosynthesis · Microbial Metabolism and Applications
Introduction
Pseudogymnoascus, a genus within the Ascomycota phylum, is renowned for its capacity to flourish in exceptionally cold conditions, categorizing it as a psychrophilic fungal group (Wang et al. 2015). While these organisms can be found worldwide, they are especially prevalent in polar areas such as the Arctic and Antarctica (Pannkuk et al. 2014). Their remarkable ability to endure and prosper in harsh environments—including low temperatures, intense ultraviolet radiation, and scarce nutrients—has made them a focal point of ecological and biotechnological research (Hassan et al. 2016). Species of Pseudogymnoascus found in Antarctica have shown the capability to produce enzymes that function effectively in cold environments, including proteases, lipases, and hydrolases. These enzymes are highly sought after for industrial applications that operate at low temperatures due to their exceptional performance in such conditions (Zucconi et al. 2020). Furthermore, Antarctic Pseudogymnoascus strains generate unique bioactive substances, such as sesquiterpenoids and polyketides, which exhibit significant antibacterial and anticancer effects. The varied fungal populations in Antarctic soils and sediments, encompassing Pseudogymnoascus species, display robust enzymatic activity and substantial biosynthetic capacity (Shi et al. 2024). This positions them as valuable targets for industrial applications and bioprospecting endeavors to uncover new bioactive molecules (Ogaki et al. 2020). Several species of the genus Pseudogymnoascus have been described in this region. Among those isolated from Antarctic soils are P. antarcticus, P. verrucosus, P. destructans, P. pannorum, and, more recently, P. russus and P. irelandiae. The species P. australis was subsequently discovered in Antarctic marine sponges. This species is characterized by unique morphological and phylogenetic traits that demonstrate its exceptional adaptation to thrive in cold environments (Villanueva et al. 2021; Childress et al. 2025). Research on P. australis has also explored its reactions to UV-B radiation stress. The fungus exhibits effective mechanisms to combat UV-induced damage, as revealed through transcriptomic analysis. This study identified the activation of crucial DNA repair pathways, including base excision repair (BER) and nucleotide excision repair (NER). These mechanisms are vital for maintaining the integrity of the fungal genome when exposed to UV radiation (Teoh et al. 2024). These findings highlight the significant biotechnological potential of these strains. However, to fully harness this potential, it will be essential to implement advanced -omics tools, such as genomics and proteomics, which enable a comprehensive understanding of biological systems by integrating data from different molecular levels (Palazzotto and Weber 2018).
Genome mining has also emerged as an innovative approach to enhance the discovery of new bioactive compounds (Albarano et al. 2020). Research indicates that Antarctic microorganisms can harbor numerous biosynthetic gene clusters (BGCs) for secondary metabolite production, with only approximately 10% of BGCs currently described (Benaud et al. 2021). Culture-dependent techniques rely on the expression of these BGCs to detect, purify, and characterize secondary metabolites. Unfortunately, many biosynthetic pathways remain dormant or cryptic under conventional culture conditions, making them inaccessible for further investigation (Scherlach and Hertweck 2009; Hoshino et al. 2019). Consequently, new methods to activate these silent gene clusters are necessary to improve the exploration and exploitation of microbial strains to discover bioactive metabolites (Ochi and Hosaka 2013). A multi-omics approach that integrates genomics and metabolomics could further enhance the identification and characterization of novel bioactive compounds (Palazzotto and Weber 2018).
Research by Khalil et al. 2014 demonstrates that nitric oxide (NO) influences the expression of BGCs in fungi and bacteria. Their findings reveal that both internally produced and externally introduced NO function as transcriptional regulators, triggering dormant BGCs in Streptomyces sp. CMB-M0423. The researchers developed novel NO delivery techniques, including in situ generation through natural chemical cues, lipopolysaccharides (LPS) from Gram-negative bacteria, and external administration via sodium nitroprusside or 1% NO in N_2_ (Khalil et al. 2014). These approaches were considered efficient and economical for accessing valuable microbial compounds.
Dewapriya et al. 2018 introduced a high-yield microbioreactor technique in 24-well plates, named MATRIX, which facilitates rapid and consistent 1 mL cultures of fungal isolates under various conditions (Dewapriya et al. 2018). This method produces numerous reference extracts for strain-specific chemical analysis and can be scaled up to preparative multi-flask/plate formats. In a separate study, Núñez-Montero et al. 2020 examined 34 Antarctic bacterial strains for the production of antibacterial secondary metabolites using LPS elicitation, sodium nitroprusside (SNP), and co-culture techniques. Employing HPLC–QTOF–MS/MS and molecular networks, they observed varying antibacterial activity depending on the strain and medium. Seven strains were chosen for genomic analysis, revealing activated biosynthetic pathways and BGCs associated with actinomycin, carotenoids, and bacillibactin. This approach, supported by genomic and metabolomic analyses, underscores the potential of these tools to unveil chemical diversity in polar microorganisms (Núñez-Montero et al. 2020). However, the systematic application of these elicitation strategies in Antarctic fungi remains underexplored. This study aims to fill that gap by investigating the potential of elicitors such as NO and LPS to activate the production of secondary metabolites in Antarctic fungi, a group with largely unknown biosynthetic potential (Xu et al. 2019). Beyond laborious traditional methods, the integration of genomic and metabolomic analyses offers an efficient pathway to navigate complex chemical spaces and directly target the discovery of novel bioactive Metabolites (Maansson et al. 2016). These metabolites have considerable biotechnological applications, including antifungal, antioxidant, and antitumor agents, as well as natural pigments, biopolymers, and other industrial products (Vaishnav and Demain 2011). Consequently, this research aims to examine the genome and identify the biosynthetic gene clusters of the Antarctic strain P. australis. Additionally, it seeks to assess the effects of NO and LPS elicitation on the metabolomic profile using HPLC–QTOF–MS/MS and molecular networking techniques.
Materials and methods
Isolation, purification, and characterization of Antarctic fungi
The study utilized samples from (Núñez-Montero et al. 2020). During Antarctic expeditions in 2014 and 2016, fungal specimens were collected from diverse sites across the Antarctic Peninsula, including soil, seawater, and sediment environments. One of the isolates, UA-032-E, was obtained from shoreline sediment at a depth of 0.3 m in the Collins Glacier area, King George Island, Antarctica (67.90665109°S, 69.16353777°E; elevation 0 m). The environmental context of this site is classified as a polar region (broad scale) and shoreline sediment (local scale). The collection date was 1 January 2016. Fungal colonies were isolated and purified through incubation on potato dextrose agar (PDA) at 15°C for up to 30 days. For subsequent experiments, inocula were prepared by cultivating each purified strain on ISP-2 and PDA plates under identical conditions (15°C, 10 days). This methodology ensured the recovery of viable and representative fungal cultures suitable for downstream analyses. The identity and purity of the cultures were confirmed through microscopic observations and genetic assessments, which included amplification and sequencing of the internal transcribed spacer (ITS) molecular marker, following the protocol described by (Pereira et al. 2023). Among the isolated fungi, the strain P. australis was selected due to evidence in the scientific literature supporting its remarkable biotechnological potential.
DNA extraction, library preparation, and whole-genome sequencing
DNA extraction was performed from a pure strain of Pseudogymnoascus australis using the DNeasy UltraClean Microbial Kit (QIAGEN, Germany) following the manufacturer’s instructions. DNA was quantified using a Qubit dsDNA HS Assay Kit (Invitrogen, USA), with a minimum concentration of ≥ 50 ng/µL. The integrity of the obtained DNA was visualized on an agarose gel in TAE 1× 0.9% m/v buffer. Samples were sequenced using Illumina and Oxford Nanopore Technologies (ONT). The Illumina library was prepared with 2 × 150 bp paired-end fragments on an Illumina NovaSeq platform. Quality control of the reads was assessed using FastQC v0.11.9 (Anon 2025). Adapters were trimmed and reads filtered using Fastp v0.20.0 (Chen et al. 2018) with the following parameters: --detect_adapter_for_pe -f 12 -F 12. For long-read sequencing, the Rapid Sequencing Kit SQK-RBK004 (ONT) was used for library preparation, and sequencing was performed using an R9.4 flow cell (FLO-MIN106D) on a MinION Mk1C machine with MinKNOW v4.3.7 software. Basecalling was performed using Guppy v5.0.12 in fast mode. Quality control was assessed using Nanoplot v1.40.0. Adapters were trimmed with Porechop v0.2.4, and sequences with a quality ≥ 10 were retained for further analysis using Nanofilt v2.8.0 (De Coster et al. 2018).
Hybrid genome assembly
Following the assembler’s command list, a hybrid assembly between Illumina paired-end and Nanopore reads was performed using MaSuRCA (https://github.com/alekseyzimin/masurca) (Zimin and Salzberg 2020). Subsequently, POLCA was used to improve consensus accuracy in the assembly using Illumina reads with the command polca.sh -a assembly.fasta-r ‘pe_R1.fa pe_R2.fa’ -t 32 -m 32G. After polishing the assembly, the SAMBA scaffolder was used to improve assembly contiguity and fill gaps using the Nanopore long reads with the command: samba.sh -r polished_assembly.fasta -q nanopore_reads.fastq.gz -t 32 (Zimin and Salzberg 2022). The assembly quality was evaluated using QUAST (https://github.com/ablab/quast), considering metrics such as N50 and the total length of the assembled genome (Gurevich et al. 2013).
Repeat masking, gene prediction, and functional and secondary metabolism annotation
Repeat masking and gene prediction were conducted using advanced tools to ensure accurate annotation, following the pipeline outlined by (Aylward et al. 2024) as a reference. RepeatModeler v2.0.1 (Flynn et al. 2020) was utilized to generate a custom library of repeat families specific to each assembly. These libraries were then applied for soft masking of the assembly (v2.1) with RepeatMasker open-4.1.7.-p1. Gene prediction was subsequently carried out on the masked assembly (v3.0) (Stanke et al. 2006) using Funannotate predict, which integrates multiple ab initio prediction tools, including Augustus v3.4.0, GeneMark-ES (Ter-Hovhannisyan et al. 2008), Glimmer v3.0.4 (Delcher et al. 2007), and SNAP (Korf 2004). In addition to these predictors, protein evidence was incorporated through BLAST alignments against the UniProt database (Bateman et al. 2023), while tRNA genes were identified using tRNAscan-SE v2.0.12 (Lowe and Eddy 1997). This comprehensive approach ensured a robust annotation of the assembly. The functional annotation of predicted genes was performed by comparing them against various annotation databases. This process involved using stand-alone tools such as InterProScan v5.72–103 (Jones et al. 2014; Blum et al. 2021), EggNOG-mapper v2.1.112 (Cantalapiedra et al. 2021), antiSMASH fungal v7.1.12 performed with relaxed detection (Blin et al. 2021), and Phobius v1.01 (Käll et al. 2007). The resulting annotations were consolidated into a single file using the Funannotate annotate pipeline, which also included comparisons of predicted proteins with the dbCAN v13.0 database of carbohydrate-active enzymes (CAZymes) (Yin et al. 2012) and the MEROPS v12.0 protease database (Rawlings et al. 2014). Additionally, the mating-type loci were identified in this genome through local BLASTn and tBLASTn searches using MAT gene or protein sequences from closely related species. To analyze the secondary metabolism, antiSMASH analysis was performed using antiSMASH version 8.0.1 with relaxed detection strictness, employing default fungal parameters for BGC prediction (Blin et al. 2025). BiG-SCAPE analysis was performed on the antiSMASH-predicted BGCs using BiG-SCAPE (v2.0.0-beta.7) with Pfam-A.hmm profiles. The run was conducted at the region level with glocal alignment, including singletons, using a similarity cutoff of 0.5 and mixed weights (Navarro-Muñoz et al. 2019). The graphical analyses and visualizations presented in this study were performed using the Python programming language (version 3.8). For the generation, customization, and export of figures, the following packages were employed: pandas (v1.3.3) for data manipulation and structuring, matplotlib (v3.4.3) as the base library for creating static plots, seaborn (v0.11.2) to enhance the aesthetic style of visualizations, and plotly (v5.3.1) for generating interactive graphs, while the circular genome visualization was performed using Circus v0.69-8 (Naquin et al. 2014).
Screening of culture conditions for elicitation of secondary metabolites
A mycelial fragment of the fungal strain, obtained from cultures grown on PDA medium, was inoculated into 1.5 mL of each of twelve different media using a micro-bioreactor system (Applikon Biotechnology, Delft, The Netherlands). An uninoculated well for each medium was kept as a negative control. Cultures were incubated at 15°C and 190 rpm for 7 days. The extraction and detection of secondary metabolites were conducted as explained in the next section. Five culture media were selected (M2, IMA, YES, ISP-4, and CG4) containing different sources of C and N as shown in Table 1 as more suitable for secondary metabolite production. The cultures were repeated using selected media as previously described under two elicitation conditions: sodium nitroprusside (SNP 2 μM) as a nitric oxide donor and LPS from Escherichia coli O55:B5 (Merck LPS 0.6 nM). In addition, sterile water or spent SNP (exposed to light for 10 days) was used as an untreated control for LPS and SNP, respectively. After 7 days of incubation at 15°C and 190 rpm, extraction and detection of secondary metabolites were performed for each treatment.
Extraction and detection of secondary metabolites by LC–MS/MS
Extraction of each culture was performed in situ by adding 2 mL of ethyl acetate (EtOAc). After [min of incubation at 190 rpm, the organic phase containing secondary metabolites was collected, dried under N_2_ airflow, and reconstituted in 20 μL of methanol for LC–QTOF (+)–MS/MS analysis. Aliquots (1 μL) of the dried resuspended fraction were analyzed on an Agilent 6545 Q-TOF LC/MS equipped with an Agilent 1290 Infinity II UPLC system. Chromatographic separation was carried out on a Zorbax C8 RRHD column (1.8 μm, 2.1 × 50 mm; Agilent). Elution used a 2.5-min linear gradient at 0.417 mL/min, from 90% water/10% acetonitrile to 100% acetonitrile, enabling efficient elution of increasingly hydrophobic analytes. Mass spectrometric detection was performed under conditions optimized to favor protonation. Under these acquisition settings, [M+H]+ adducts consistently represented the most abundant and reproducible features across all samples. Downstream analyses therefore focused on [M+H]+ ions to ensure consistency and comparability among strains. A collision energy of 20 eV was applied to induce fragmentation of precursor ions and generate product ion spectra suitable for structural elucidation.
LC–QTOF–MS/MS metadata analysis and annotation of secondary metabolites
The MZmine v4.0.3 program (Kim et al. 2021) was used to preprocess the relevant adduct ions [M+H]+ for Feature-Based Molecular Networking (FBMN). The parameters were kept for all strains processed and are as follows: Ion Mode = Positive Absolute Intensity; Noise Threshold = MS1: 1.0E5, MS2: 1.0E2; Minimum Feature Height = 3.0E2; Crop Retention Time = autorange; Feature list blank subtraction of the spectra of the media and blanks used. The molecular networks were created using the GNPS FBMN analysis and annotation tool (Wang et al. 2016) for all strains. The following parameters were kept consistent: Precursor Ion Mass Tolerance = 0.02; Fragment Ion Mass Tolerance = 0.02. All datasets used in this project are available in the GNPS repository: ftp://massive.ucsd.edu/MSV000085261/. The SIRIUS (Dührkop et al. 2019) program tools were utilized to perform in silico metabolite prediction and annotation. SIRIUS, ZODIAC, CSI:FingerID, and CANOPUS tools were used for this process. For CSI:FingerID, all available databases were selected for compound identification, and CANOPUS was used for natural product class predictions according to NPClassifier (Kim et al. 2021). Visualization and data processing were done on Cytoscape v3.7.1 (The Cytoscape Consortium, California, USA) and included the data from both GNPS FBMN annotation and SIRIUS in silico predictions to facilitate the analysis of all features.
Results
Sequencing and annotation of the Pseudogymnoascus
australis UA-032-E
The complete genome of the fungal strain P. australis UA-032-E was sequenced using Nanopore and Illumina technologies, resulting in a hybrid assembly. The resulting sequences were evaluated according to quality parameters, and it was determined that the genome has over 96% completeness. Additionally, the genome assembly consists of 33 contigs, with the largest contig measuring 3,190,541 bp and a total genome length of 37,254,395 bp (Fig. 1). The genome assembly of Pseudogymnoascus australis UA-032-E demonstrates high continuity, as reflected by an N50 of 1.91 Mbp and an N90 of 0.97 Mbp, with L50 and L90 values of 7 and 18, respectively. The overall GC content was 50.16%. The assembled genome has been deposited in the NCBI database under the accession number JBLVTM000000000. Phylogenetic analysis based on concatenated ITS, RPB2, and Tef1 sequences strongly supports the classification of UA-032-E within the P. australis clade, with high statistical support (99% ML bootstrap / – BI posterior probability), clearly distinguishing it from related species such as P. griseus, P. papyriferae, and P. shaanxiensis (Suppl. material 1: section S1). Functional annotation identified 10,800 protein-coding genes, along with 48 rRNA and 199 tRNA genes. Additionally, 2,923 proteins were predicted to contain transmembrane domains, and 9,981 proteins were associated with PFAM domains, highlighting the functional diversity and genomic completeness of this Antarctic fungal strain.
Genome assembly visualization of P. australis UA-032-E using a Circus plot. The image depicts the genomic structure and organization, comprising 33 contigs. Colors on the outer ring denote genes with unidentified functional elements within the genome, according to the legend. Functional classification of genes was conducted using the Clusters of Orthologous Groups (COGs) database categories, thus offering a comprehensive view of the genome architecture and organization of P. australis UA-032-E. Circular representation was done using Circos v0.69-8.
Analysis of biosynthetic gene clusters of the P.
australis UA-032-E
The genome analysis of P. australis UA-032-E using the antiSMASH tool led to the prediction of a total of 44 BGCs. Among them, 13 BGCs showed significant similarity to previously characterized entries in the MIBiG database, based on their BLAST score, percentage identity, coverage, and E-value. As shown in Fig. 2, which graphically represents the identity and coverage values of each annotated BGC, those located in the upper right quadrant meet both threshold criteria (≥50% identity and ≥90% coverage), indicating a high degree of functional similarity with reference BGCs. Notable examples (Table 2) include clusters associated with the production of geodin (BGC0002592.2; 70% identity, 99.39% coverage), F9775A/F9775B/orsellinic acid (BGC0000057.5; 74%, 99.70%), and ustilaginoidin N/O/M/A (BGC0002177.2; 66%, 93.23%). Conversely, BGCs such as BGC0000037.4 (cichorine) and BGC0002175.3 (YWA1) exhibited coverage values exceeding 100% (103.18% and 102.37%, respectively) but only moderate identity (46% and 48%), suggesting the presence of structurally conserved variants with sequence divergence. In contrast, BGC0001839.3 (squalestatin S1) showed high identity (70%) but low coverage (56.16%), placing it outside the upper threshold.
Comparative analysis of BGCs identified in P. australis UA-032-E using antiSMASH and annotated by similarity to the MIBiG database. The X-axis shows sequence identity (%) and the Y-axis shows coverage (%) based on BLAST alignments between the predicted BGCs and their most similar known counterparts in MIBiG. Each point represents an individual BGC, where the color indicates the biosynthetic type (e.g., PKS, NRPS, terpene, or hybrids), the point size corresponds to the BLAST score (larger size = stronger similarity), and the symbol shape reflects the confidence level assigned to the annotation. Dashed lines represent selection thresholds: 50% identity (vertical turquoise line) and 90% coverage (horizontal red line), which are considered the minimum criteria to classify a BGC as highly similar to a previously characterized cluster. BGCs located in the upper right quadrant exhibit both high identity and high coverage, suggesting strong functional similarity to known clusters.
Table 2.: BGCs identified in P. australis UA-032-E using antiSMASH and their similarity to MIBiG entries.
Following the identification of 44 BGCs in the genome of P. australis UA-032-E using antiSMASH, comparative analysis was performed using BiG-SCAPE to group them into functional families based on structural and sequence similarities. This large-scale comparative approach allows for the inference of evolutionary relationships and the identification of potentially novel biosynthetic pathways, facilitating the discovery of bioactive secondary metabolites. The predicted BGCs encompassed a broad range of biosynthetic types: T1PKS, 20 clusters, followed by NRPS, 8 clusters, terpenes, 5 clusters, and several hybrid or variant types, including NRPS-T1PKS, 4 clusters (Fig. 3a). The analysis revealed a total of 41 gene cluster families, of which 39 were singletons, indicating that most BGCs shared low similarity with each other and likely represent unique biosynthetic pathways (Fig. 3b). Two multi-member BGC families, FAM_00001 and FAM_00002, suggest minimal genetic redundancy in P. australis, indicating a limited biosynthetic repertoire (Suppl. material 1: section S2). Notably, the BGC family FAM_00002 displays a unique genomic arrangement, with identical assemblies found on three separate contigs. This family comprises three NRPS-type BGCs with a modular structure featuring the canonical A–T–C domain triad, which includes PF00501 (AMP-binding; adenylation domain), PF00550 (phosphopantetheine attachment site; thiolation domain), and PF00668 (condensation domain). This configuration is characteristic of NRPS systems that facilitate substrate activation and peptide bond formation. These clusters are predicted to produce small cyclic peptides with 2 to 4 residues, incorporating various combinations of aromatic (e.g., phenylalanine, tyrosine, tryptophan) and aliphatic (e.g., valine, isoleucine) amino acids. Interestingly, two of these NRPS closely resemble BGC0000357.5 from Penicillium rubens, a known cluster encoding diketopiperazine synthases that generate cyclopeptides with the general structure cyclo-(D-aa–L-aa–D-aa–L-aa). Conversely, the BGC family FAM_00001, with identical assemblies on two distinct contigs (scaffold_17.region002 and scaffold_13.region001), encodes a highly conserved iterative T1PKS involved in synthesizing 1,3,6,8-tetrahydroxynaphthalene (THN), a crucial precursor in the DHN-melanin biosynthetic pathway. Comparative analysis using the MIBiG database reveals that FAM_00001 shares 81% sequence identity with BGC0001258.3 from Glarea lozoyensis, a known THN-producing cluster. Functional annotation of its biosynthetic genes shows a minimal yet catalytically complete PKS module, including PF00109 (β-ketoacyl synthase N-terminal domain), PF00083 (β-ketoacyl synthase C-terminal domain), PF00698 (acyltransferase), PF16073 (ACP transacylase), PF00550 (phosphopantetheine attachment site; ACP), PF14765 (dehydratase domain), and PF00975 (thioesterase domain). This setup supports iterative polyketide chain elongation through decarboxylative condensations of malonyl-CoA units (facilitated by the AT domain with a conserved GQGxQ motif), intermediate shuttling by the ACP, and product release by the TE domain, ultimately leading to the formation of the naphthalene scaffold through spontaneous cyclization.
Comprehensive analysis of BGC distribution in the genome of P. australis UA-032-E. a Distribution of dominant BGC types across scaffolds, revealing the predominance of T1PKS systems. Color coding represents biosynthetic types: NRPS (blue), T1PKS (light blue), PKS-NRPS hybrids (light green), NRPS-like (green), T3PKS (dark green), and terpene (red). Bar values indicate the number of scaffolds and corresponding percentage. The dashed red line marks the average number of scaffolds per type. This distribution suggests specialized genomic regions favoring particular biosynthetic pathways with significant representation of both modular systems and specialized metabolism, highlighting the functional organization of the organism’s biosynthetic potential. b Heatmap displaying BGCs grouped by family (vertical axis) and their genomic location by scaffold (horizontal axis). BiG-SCAPE analysis identified 44 BGCs classified into 41 families, with only two families containing multiple members, indicating high structural diversity.
Metabolomic profiling and structural elucidation of compounds from P.
australis UA-032-E under varied growth conditions
Metabolomic profiling of P. australis was performed using LC–QTOF–MS/MS, with subsequent data analysis leveraging the GNPS Feature-Based Molecular Networking platform and the SIRIUS/CANOPUS computational suite. This integrated strategy enabled extensive annotation of biologically relevant molecular features by combining spectral matching with in silico fragmentation prediction. A Venn diagram (Fig. 4a) illustrates the overlap of metabolic features across the four experimental treatments [LPS(−), LPS(+), NPS(−), NPS(+)], revealing a core set of 75 MS features consistently present under all conditions. Results are interpreted at the level of putatively annotated MS features rather than fully validated metabolites. Further analysis of feature distribution across culture media (CGA, IMA, ISP-4, M2, YES) and elicitation conditions [NPS(−), NPS(+), LPS(−), LPS(+), and combined NPS(+)/LPS(+)] is presented in a bar chart (Fig. 4b). The combined elicitation treatment (NPS+/LPS+) yielded the highest number of detected features. Among culture media, YES and CGA supported the greatest diversity of secondary metabolite-related features, indicating their particular suitability for eliciting a broad spectrum of metabolic responses in P. australis.
Venn diagram showing the distribution of compounds identified in the treatments LPS(−), LPS(+), NPS(−), and NPS(+). a A common core of 75 features is present across all four treatments, while other areas of the diagram reveal compounds specific to each treatment or combination of them. Positive treatments (LPS+ and NPS+) share a greater number of compounds with each other compared to the negative treatments, suggesting potential differences in the metabolic responses induced by the experimental stimuli. b The graph represents the number of features generated with respect to the growth medium and the treatment applied to the samples. A stacked bar chart was used to visualize these variations across six distinct media: CGA, ES, IMA, ISP-4, M2, and YES. The treatments evaluated included combinations of NPS(−), NPS(+), LPS(−), LPS(+), and NPS(+)/LPS(+).
Metabolic profile and diversity of secondary metabolites in P.
australis UA-032-E
The chemical composition of P. australis was characterized through a comprehensive metabolomic analysis employing LC–QTOF–MS/MS. Compound annotation was achieved by integrating two complementary computational approaches. First, molecular networking via the GNPS platform facilitated the dereplication of known metabolites by comparing acquired MS/MS spectra against public spectral libraries and grouping related molecules based on spectral similarity (Fig. 5). Second, to annotate novel or library-unmatched features, the SIRIUS computational suite was used, which combines CSI:FingerID for molecular structure prediction via fragmentation tree analysis and CANOPUS for predicting compound class labels directly from MS/MS spectra without requiring a reference library. This combined strategy allowed for the comprehensive annotation of both known and previously unreported metabolites in the extract.
Molecular networking of adduct ions produced by P. australis UA-032-E cultivated under different conditions, analyzed using HPLC–QTOF–MS/MS, with metadata processed through the GNPS platform and SIRIUS/CANOPUS. Border color refers to the metabolic pathway predicted by CANOPUS: green for alkaloids, yellow for polyketides, blue for amino acids and peptides, red for shikimates and phenylpropanoids, burgundy for fatty acids, pink for terpenoids, purple for carbohydrates, and white for not identified. Fill colors represent the elicitation treatment at culture: blue for LPS−, grey for LPS+, yellow for NPS−, orange for NPS+, and green for multiple treatments excluding LPS−. Ring chart colors refer to the culture medium used: red for CGA, orange for ES, light orange for IMA, yellow for ISP-4, green for M2, and blue for YES. The diameter of the nodes/features is proportional in size to the intensity of the precursor in the initial sample. The width of the edges represents the cosine score with values between 0.5 and 5.0.
The GNPS molecular network revealed a significant presence of different metabolites (Table 3). Notably, diacylglyceryl-N,N,N-trimethylhomoserine (DGTS 18:2) was identified, producing a characteristic fragment ion at m/z 498.3800. This fragment corresponds to the monoacyl form, comprising a glycerol backbone, homoserine, and a single linoleic acid (C18:2) chain, which is consistent with the established fragmentation pattern for DGTS-type betaine lipids. Furthermore, the alkaloid harman was annotated and detected as its protonated adduct [M+H]^+^ at m/z 183.0910. The high spectral similarity to previously reported data supports its confident identification and indicates the presence of β-carboline alkaloids in the metabolic profile of this fungus. Another identified compound was N^6^,N^6^-dimethyladenine (N,N-dimethyl-7H-purin-6-amine), observed as a protonated molecule [M+H]^+^ at m/z 164.0930, matching its exact theoretical mass. This methylated purine derivative is a known precursor and has been implicated in fungal cytokinin biosynthesis pathways. Recent studies have established that certain fungal species, including Claviceps purpurea, biosynthesize bioactive cytokinins, such as trans-zeatin, via dedicated de novo pathways (Hinsch et al. 2015). Furthermore, three diketopiperazine (DKP) cyclic dipeptides were annotated based on precursor masses matching their theoretical values in [M+H]^+^ mode. The compounds cyclo(L-Phe-L-Pro) (m/z 245.1280), cyclo(L-Val-L-Pro) (m/z 197.1281), and cyclo(L-Pro-L-Leu) (m/z 211.1437) were identified. These metabolites, characterized by their stable diketopiperazine ring structure, are commonly produced by fungi. Their annotation is corroborated by the GNPS molecular network, where each compound formed a distinct node. The identification of cyclo(L-Val-L-Pro) is consistent with prior reports from fungi such as Penicillium chrysogenum and Schizophyllum commune. Similarly, cyclo(L-Pro-L-Leu) has been previously isolated from various fungal species, including the endophytic fungus Aspergillus terreus and marine-derived Pseudofusicoccum sp. (Bach et al. 2022). Furthermore, the cyclic dipeptide cyclo(L-Pro-L-Leu) was identified with an observed [M+H]^+^ ion at m/z 211.1430. This diketopiperazine has been previously reported in various fungi, including the endophytic strain Aspergillus terreus GX-3 and the marine-derived fungus Pseudofusicoccum sp., often co-occurring with other bioactive DKPs (Ma et al. 2021; Alves et al. 2025). The presence of cyclo(L-Pro-L-Leu), along with cyclo(L-Phe-L-Pro) and cyclo(L-Val-L-Pro), is robustly supported by the GNPS molecular network, wherein each metabolite forms a distinct node connected by spectral similarity to known library matches. This consolidated evidence confirms the production of these characteristic fungal diketopiperazines by P. australis.
Table 3.: Metabolites annotated via GNPS from P. australis under different treatments.
Annotations generated by the SIRIUS platform further highlighted a diverse array of peptide and polyketide derivatives (Table 4). Notably, the cyclic diketopiperazine maculosin [cyclo(L-Pro-L-Tyr)] was identified, with an observed [M+H]^+^ ion at m/z 261.1230, matching its exact theoretical mass. This compound, previously isolated from fungi such as Alternaria alternata, is recognized for its antimicrobial properties. Additionally, the polyketide-derived metabolite methyl asterrate was annotated among the detected secondary metabolites (Castaldi et al. 2022). The methyl ester derivative methyl asterrate was tentatively annotated based on an observed [M+H]^+^ ion at m/z 385.0896. This fungal secondary metabolite, previously reported in Geomyces sp., has been associated with inhibitory activity against Aspergillus fumigatus (Liu et al. 2016). Similarly, the oxygenated xanthone derivative 3-hydroxy-1,2,4-trimethoxyxanthen-9-one was identified with a precursor mass of m/z 303.0861 [M+H]^+^, matching its theoretical mass. This class of compounds is commonly produced by fungi, including genera such as Penicillium, Xylaria, and Paraphaeosphaeria (Ge et al. 2006; Shao et al. 2008; Wang et al. 2018).
Table 4.: Metabolites annotated via SIRIUS from P. australis under different treatments.
Discussion
The fungal genus Pseudogymnoascus is renowned for its remarkable ability to thrive in extreme conditions, particularly in polar regions characterized by low temperatures, intense ultraviolet radiation, and limited nutrients. These demanding environments have fostered the evolution of unique metabolic and physiological adaptations in these organisms. Recent studies have demonstrated that Pseudogymnoascus produces secondary metabolites with considerable biotechnological applications, including pharmaceutical and textile use (Satriawan et al. 2024). A notable example is the research conducted by Antipova et al. (Antipova et al. 2023), which explored the genus Pseudogymnoascus and uncovered its exceptional biosynthetic capacity for generating bioactive compounds. Their investigation identified the production of 16-membered trilactone macrolides, (+)-macrosphelides A and B, in the Arctic strains VKM F-4518 and VKM F-4519, with macrosphelide A emerging as a promising candidate for cancer therapy. Moreover, a comprehensive analysis of metabolites produced by the fungus Pseudogymnoascus sp. HSX2#-11, guided by molecular networking techniques, resulted in the isolation of a new polyketide, pseudophenone A, along with six previously identified analogues. The structure of this novel compound was elucidated through extensive spectroscopic analysis and single-crystal X-ray diffraction. The compound, named pseudophenone A, is composed of a dimer of diphenyl ketone and diphenyl ether, and to our knowledge, only one analog of this structural class has been previously reported. Both pseudophenone A and compound 2 displayed antibacterial activity against various bacterial strains (Shi et al. 2021). These discoveries emphasize the effectiveness of molecular networking as a powerful tool for identifying new bioactive compounds in psychrophilic fungi, thus supporting the methodology employed in our study. Furthermore, they underscore the substantial metabolic potential of Pseudogymnoascus spp. as a valuable resource for future investigations aimed at exploring secondary metabolites with biotechnological applications (Yoshinaga et al. 2024).
P. australis, a species discovered in Antarctic marine sponges, exhibits distinct morphological and phylogenetic traits that set it apart from other members of its genus (Villanueva et al. 2021). Teoh et al. (Teoh et al. 2024) conducted a study examining the transcriptomic responses of an Arctic strain to UV-B radiation. The strains were grown on nylon membranes atop Czapek-Dox agar at 15°C in dark conditions for 10 days until reaching exponential phase. They were then subjected to a UV-B dose of 6.1 kJ m^−2^ d^−1^ for 130 minutes and examined under light after 0, 2, and 6 hours. RNA-Seq analysis (40 million reads per sample) revealed an increase in radiation repair (RAD) genes and base excision repair (BER) and nucleotide excision repair (NER) genes. In contrast, the photoreactivation gene (PHR1) showed decreased expression. Additionally, a reduction in oxidoreductase activity was observed as a stress adaptation mechanism. This pioneering analysis sheds light on P. australis DNA repair mechanisms, offering insights into its resilience to extreme environments and UV-induced damage. The complete genome sequencing of P. australis marks a significant advancement in our comprehension of its genetic and enzymatic potential. This achievement enables the precise identification of crucial genes and exploration of related biosynthetic pathways, underscoring the organism’s substantial biotechnological value.
To date, only one study has reported the identification of secondary metabolites in P. australis from the Arctic region using the antiSMASH tool. Antipova et al. (Antipova et al. 2023) reported 32 BGCs in the genome of strain VKM F-4518, including 11 T1PKS, 9 NRPS, 1 T3PKS, 1 mixed (T1PKS, NRPS), 7 fungal RiPP, and 3 terpene cyclases (T). In strain VKM F-4519, 17 clusters were identified: 8 T1PKS, 6 NRPS, 1 hybrid (T3PKS and NRPS), and 2 fungal RiPP, with similarities to known pathways ranging from 12% to 100%. The genomic analysis of P. australis UA-032-E identified 44 BGCs, of which 13 showed high similarity to known clusters in MIBiG (e.g., geodin, ustilaginoidins). Among them, the most notable are 6 PKS, 2 iterative type I PKS, 1 type I NRPS, 2 NRPS–PKS hybrid clusters, 1 classified as other, 1 terpene, and 1 hemiterpene cluster. When comparing P. australis from Arctic regions with the Antarctic-origin P. australis from our study, both exhibit a clear tendency to produce PKS- and NRPS-type BGCs. The high abundance of PKS and NRPSBGCs in fungi reflects their evolutionary adaptation to diverse ecological niches, enabling the production of complex secondary metabolites for defense, competition, and symbiosis (Schümann and Hertweck 2007; Minami et al. 2020; Allen et al. 2024). This metabolic diversity is further shaped by gene duplication, environmental pressures, and host interactions (Bergmann et al. 2007; Collemare et al. 2008).
BiG-SCAPE analysis, used to group BGCs based on structural and functional similarity, identified two notable GCFs: FAM_00002, predicted to encode an NRPS for cyclic peptide biosynthesis, and FAM_00001, an iterative PKS putatively involved in THN biosynthesis. The remaining 39 BGCs did not cluster into families (i.e., were singletons), a common outcome reflecting high biosynthetic diversity. This pattern of few conserved families alongside abundant singletons suggests that, like other fungal species such as Xylaria hypoxylon and Aspergillus spp., P. australis possesses a reservoir of unique biosynthetic potential. This significantly expands the known chemical space available for the discovery of novel bioactive secondary metabolites (Yang et al. 2020). Therefore, beyond characterizing genomic diversity, BiG-SCAPE is instrumental in prioritizing novel clusters, particularly those within GCFs, for functional studies or drug discovery efforts.
Metabolomic analysis and elicitation treatments using various media and substances like LPS and SNP resulted in the induction of 75 features, which were identified through LC–QTOF–MS/MS. This outcome demonstrates the efficacy of this approach in generating new compounds. A study by Khalil et al. (Khalil et al. 2014) demonstrated that LPS serves as a natural chemical stimulant capable of altering secondary metabolic expression in fungal species such as Penicillium sp. and Aspergillus sp. The research indicated that LPS treatment activated secondary metabolites in 15% of the studied strains, with an optimal concentration of 0.6 ng/mL. Additionally, this activation enhanced the production of bioactive substances, including pseurotins and neoasterriquinones. These findings indicate that LPS stimulation can unlock the molecular potential of fungal strains previously inactive under standard cultivation conditions. Research on NO has shown its considerable potential and identified several crucial mechanisms underlying its effects. NO functions as a signaling molecule regulating vital processes in fungi, with nitrate reductase producing it in response to environmental stressors or during fungal developmental phases. Moreover, NO plays a critical role in neutralizing nitrosative stress and promoting biosynthetic pathways for secondary metabolites, thereby enhancing the expression of genes involved in producing cryptic metabolites (Cánovas et al. 2016; Zhao et al. 2020). The set of 75 synthesized features displays a broad range of structural variations. The use of LPS and SNP elicitors together, irrespective of the culture medium, resulted in a greater production of compounds than when each was used separately (Fig. 3a). These results are consistent with those of Núñez-Montero et al. (Núñez-Montero et al. 2020), who found through MS/MS metabolomics and genome mining that combining elicitors can trigger dormant metabolic pathways and enhance metabolite diversity in Antarctic bacteria. The synthesis of these molecules can be influenced by various environmental conditions and the presence of elicitors that trigger specific metabolic pathways. Biotic elicitors, such as fungi and bacteria, have been identified as key agents in the induction of secondary metabolite biosynthesis. Bhaskar et al. (Bhaskar et al. 2021) reported that exposure to Aspergillus niger increased thiophene production by 85%, while Botrytis spp. promoted a 26-fold increase in sanguinarine synthesis. Additionally, extracts from Haematococcus pluvialis enhanced betalain production by 2.28-fold. Regarding chemical elicitors, methyl jasmonate (MeJA) has been identified as a potent regulator of secondary metabolite synthesis, with reports indicating up to a 2230-fold increase in fungal cultures (Giri and Zaheer 2016). Beyond enhancing the biosynthesis of previously characterized compounds, elicitors can activate new metabolic pathways. Alghuthaymi et al. (Alghuthaymi et al. 2022) demonstrated that MeJA treatment significantly increased rosmarinic acid accumulation (117 mg/g dry weight) in Halodule pinifolia cultures, along with the synthesis of 47 additional metabolites, including flavonoids and phenolic acids. These findings suggest that elicitors modulate metabolic production and trigger the synthesis of previously undetected compounds, expanding the repertoire of metabolites with potential biotechnological applications. The chemical detection of cyclic peptides [e.g., maculosin, cyclo(-Pro-Val)] in the fungal extracts via LC–MS/MS confirms the activity of this specific biosynthetic machinery. The presence of these types of compounds, which are known to mediate ecological interactions and exhibit notable bioactive properties in other fungi, suggests a potential role in defense mechanisms or ecological adaptation for this strain. For example, griseaketides produced by Penicillium spp. have shown antibacterial and antifungal activity, indicating a defensive role in their natural environment (Valente et al. 2020). Co-culture experiments and stress exposure have been shown to induce the production of antimicrobial cyclic peptides in phytopathogenic fungi (Soledade and Chumala 2006), while in endophytic fungi, these metabolites have been linked to microbial competition within plant tissues (Zhou et al. 2011). The detection of the betaine lipid DGTS (18:2) suggests a potential role in membrane remodeling. This finding is intriguing given that in other organisms like Chlorella kessleri, specific DGTS species are known to replace phosphatidylcholine under phosphorus-deficient conditions as part of a strategy to maintain membrane integrity. While the detection of DGTS (18:2) in our system is consistent with a possible adaptive response to stress, a direct causal relationship in this specific context remains to be established through future targeted lipidomic studies under controlled conditions (Riekhof et al. 2014). Similar findings have been reported in fungi, where phosphate starvation triggers the replacement of phospholipids with DGTS, highlighting its conserved role in stress adaptation. Furthermore, the detection of glycosylated flavonoids, such as 7-O-methyleriodictyol 3’’-O-glucoside, is of interest due to the documented pharmacological properties of this class of compounds. For instance, studies in other systems have shown that glycosylation can enhance flavonoid solubility and stability (Krawczyk‐łebek et al. 2022), which may improve their antioxidant capacity. This is supported by work such as that of Liu et al. (Liu et al. 2022), where the induction of glycosylated flavonoids under stress was correlated with reduced oxidative damage. Thus, the presence of these metabolites in our system warrants further investigation to assess their actual antioxidant and pharmacological potential (Behr et al. 2020).
The integration of genomic and metabolomic data is essential for understanding the biosynthetic potential of an organism. Three cyclic peptides, cyclo(Pro-Val), cyclo(L-Phe-D-Pro), and cyclo(L-Pro-L-Leu), were detected through LC–QTOF–MS/MS metabolomic analysis, annotated via GNPS, and classified as amino acid and peptide derivatives by CANOPUS (Toghueo et al. 2020). These compounds were primarily induced under LPS treatment and expressed in culture media such as YES, IMA, and ISP-4. Additionally, genomic analysis using antiSMASH and BGC clustering via BiG-SCAPE revealed NRPS-type clusters with high similarity to known BGCs from the MIBiG database, supporting the hypothesis that these metabolites originate from biosynthetic gene clusters within P. australis. However, despite the use of elicitors such as NO and LPS, metabolites for some predicted BGCs were not detected. This could indicate a requirement for more specific activation signals, or it could suggest that novel metabolites were produced but remained among the unannotated features in our metabolomic datasets, highlighting the potential for discovering new chemistry (Pfannenstiel and Keller 2019). Additionally, certain clusters may be epigenetically regulated, produce metabolites at very low concentrations, or generate unstable or non-ionizable compounds under the applied analytical conditions (Robey et al. 2020; Xue et al. 2023). Tools such as GNPS and SIRIUS provide predictive metabolomic analyses, aiding in metabolite annotation and structural classification. Nevertheless, these platforms rely on mass spectrometry-based predictions; thus, further validation is required using techniques capable of elucidating the precise chemical structure, such as NMR or X-ray crystallography. These findings highlight the need to combine chemical elicitation with transcriptomic strategies and structural analytical methods to fully access the organism’s metabolic repertoire (Clevenger et al. 2017).
These observations highlight the necessity of integrated approaches that link genetic pathways to the production of novel compounds. Exploiting the potential of microorganisms to uncover new molecules could be vital in addressing the global demand for developing safer, more cost-effective, and efficient drugs for various diseases. The present moment offers an ideal opportunity to reinvigorate and intensify our dedication to microorganism-inspired drug discovery.
Conclusion
This study reports the first annotated genome of P. australis UA-032-E, revealing its considerable biosynthetic potential through the identification of 44 BGCs using antiSMASH, including NRPS, type I PKS, and hybrid systems. Analysis with BiG-SCAPE grouped these BGCs into 41 families, most of which were singletons, suggesting low redundancy and high structural diversity. The application of elicitors such as SNP and LPS in different culture media enabled the activation of previously undetected metabolic pathways. Metabolomic analysis via HPLC–QTOF–MS/MS, complemented with GNPS and SIRIUS, allowed the detection of 75 features. Among them, compounds such as cyclodipeptides [cyclo-(Pro-Val), cyclo-(Leu-Leu)], maculosin, and betaine lipids like DGTS 18:2 were identified, which have been associated in the literature with potential roles in stress adaptation or biological activities. The YES + LPS condition showed the highest metabolite diversity, indicating that this combination of medium and elicitor may be favorable for the expression of specialized metabolism. Together, these results highlight the usefulness of integrating genomic and metabolomic approaches to explore the chemical potential of psychrophilic fungi. The genomic characterization presented here constitutes a fundamental resource for future functional studies and bioprospecting efforts aimed at elucidating biosynthetic pathways and characterizing metabolites with potential biotechnological applications.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Albarano L, Esposito R, Ruocco N et al. (2020) Genome mining as new challenge in natural products discovery. Marine Drugs 18: 199. 10.3390/md 18040199 PMC 723028632283638 · doi ↗ · pubmed ↗
- 2Alghuthaymi MA, Danaraj J, Albarakaty FM et al. (2022) Elicitor-Induced Metabolomics Analysis of Halodule pinifolia Suspension Culture for an Alternative Antifungal Screening Approach against Candida albicans. Journal of Fungi 8: 609. 10.3390/jof 8060609 PMC 922478535736092 · doi ↗ · pubmed ↗
- 3Allen BM, Drott M, Nickles GR et al. (2024) Variation in Biosynthetic Gene Clusters Among Lifestyles Across Kingdom Fungi. bio Rxiv. 10.1101/2024.09.14.613087 · doi ↗
- 4Alves BVB, Borges LJ, Hanna SA et al. (2025) Pigment production by Pseudofusicoccum sp.: Extract production, cytotoxicity activity, and diketopiperazines identified. Microorganisms 13: 277. 10.3390/microorganisms 13020277 PMC 1185756140005644 · doi ↗ · pubmed ↗
- 5Anon (2025) Anon Git Hub - s-andrews/Fast QC: A quality control analysis tool for high throughput sequencing data. [Available from:] https://github.com/s-andrews/Fast QC [February 27, 2025]
- 6Antipova TV, Zaitsev KV, Zhelifonova VP et al. (2023) The potential of arctic Pseudogymnoascus fungi in the biosynthesis of natural products. Fermentation 9: 702. 10.3390/fermentation 9080702 · doi ↗
- 7Aylward J, Wilson AM, Visagie CM et al. (2024) IMA Genome – F 19. IMA Fungus 15: 12. 10.1186/s 43008-024-00142-z PMC 1114938038831329 · doi ↗ · pubmed ↗
- 8Bach TNQ, Cao DT, Hoang THL et al. (2022) Cyclodipeptides isolated from a marine-derived fungus Penicillium chrysogenum M 612 of Bai Tu Long Sea, Quang Ninh, Vietnam. IFMBE Proceedings 85: 527–537. 10.1007/978-3-030-75506-5_45 · doi ↗