ERGA-BGE Genome of Ailoscolex lacteospumosus Bouché, 1969 – the enigmatic milky worm endemic to the Pyrenees
Marta Novo, Daniel Fernández Marchán, Sylvain Gérard, Alejandro Martínez Navarro, Rita Monteiro, Astrid Böhne, Thomas Marcussen, Torsten H Struck, Rebekah A Oomen, Laura Aguilera, Marta Gut, Francisco Câmara Ferreira, Jèssica Gómez-Garrido, Fernando Cruz, Tyler Alioto

TL;DR
This paper presents the genome of Ailoscolex lacteospumosus, a rare earthworm species from the Pyrenees, to study its evolution and conservation.
Contribution
The paper provides a high-quality chromosome-level genome assembly for a rare and evolutionarily significant earthworm species.
Findings
The genome assembly includes 17 contiguous chromosomal pseudomolecules totaling 493 Mb.
The assembly has high contig and scaffold N50 values of 15.1 Mb and 29.9 Mb, respectively.
The genome will aid in understanding the evolution and conservation of this endemic earthworm species.
Abstract
Ailoscolex lacteospumosus represents one of the basal taxa of the earthworm family Hormogastridae and was initially placed in a different family, Ailoscolecidae, including just this species. Its genome can help unravel the evolution of this earthworm family, which is endemic to the Mediterranean and adapted to soils exposed to adverse conditions such as prolonged droughts. This species presents a very restricted distribution, endemic to the Pyrenees, and the genome will help study its conservation status as well as provide insights into earthworm evolution. A total of 17 contiguous chromosomal pseudomolecules were assembled from the genome sequence. This chromosome-level assembly encompasses 493 Mb, composed of 46 contigs and 24 scaffolds, with contig and scaffold N50 values of 15.1 Mb and 29.9 Mb, respectively.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
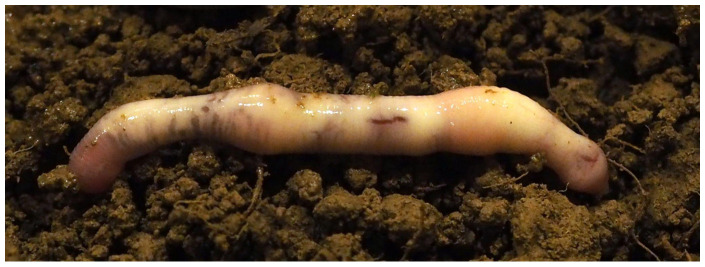 Figure 1
Figure 1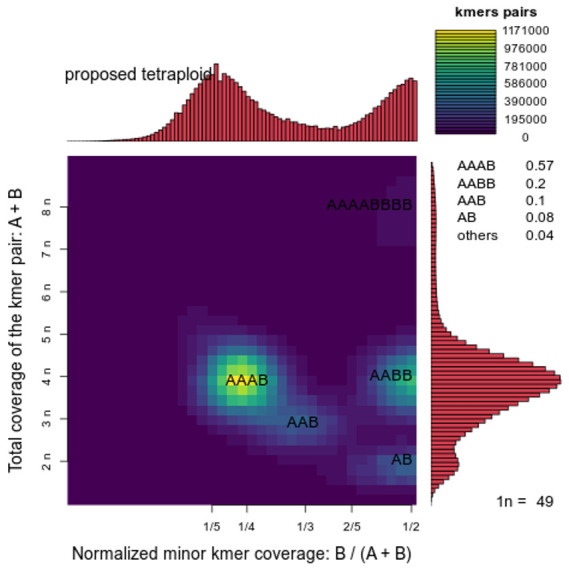 Figure 2
Figure 2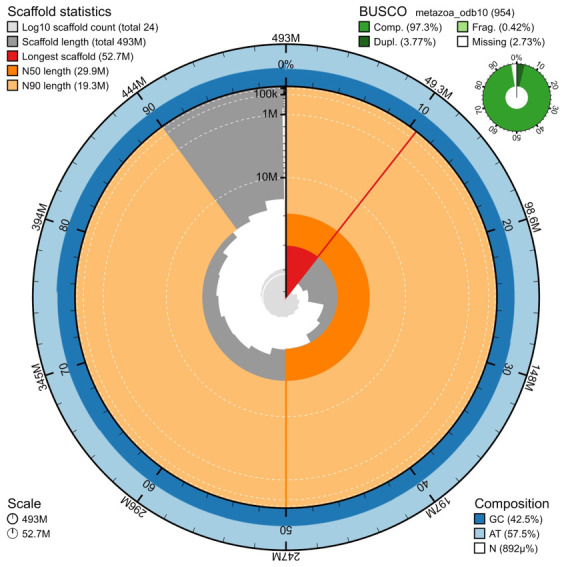 Figure 3
Figure 3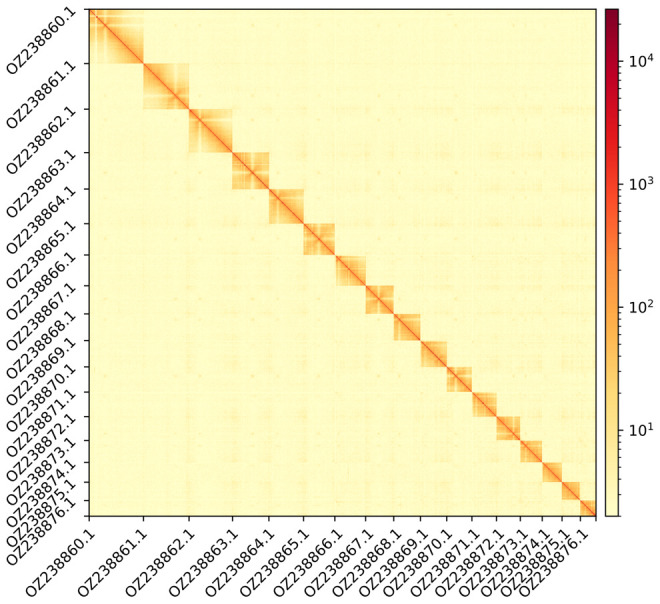 Figure 4
Figure 4| No. genes | No. transcripts | Mean
| No. single-exon genes | Mean
| |
|---|---|---|---|---|---|
|
| 23,944 | 40,974 | 11,551 | 642 | 8.7 |
|
| 3,032 | 3,482 | 4,980 | 2 | 2.4 |
|
| 64 | 64 | 156 | 64 | 1.0 |
|
| 46 | 46 | 115 | 46 | 1.0 |
|
| 25 | 25 | 119 | 25 | 1.0 |
|
| 224 | 224 | 76 | 224 | 1.0 |
|
| 2 | 2 | 130 | 2 | 1.0 |
|
| 876 | 876 | 74-87 | 876 | 1.0 |
| Complete | Singular | Duplicated | Fragmented | Missing | |
|---|---|---|---|---|---|
|
| 912 (95.6%) | 874 (91.6%) | 38 (4.0%) | 19 (2.0%) | 23 (2.4%) |
|
| 2,062 (95.7%) | 1,706 (79.2%) | 356 (16.5%) | - | 91 (4.3%) |
| Consistent | Inconsistent | Contaminants | Unknown | ||
|
| 14,784 (61.7%) | 3,066 (12.8%) | 0 | 6,094 (25.5%) | |
- —Horizon Europe Framework Programme
- —MCIN/AEI/10.13039/501100011033/FEDER, UE
- —UK Research and Innovation (UKRI)
- —Swiss State Secretariat for Education, Research and Innovation (SERI)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInvertebrate Taxonomy and Ecology · Leech Biology and Applications · Genomics and Phylogenetic Studies
Introduction
Ailoscolex lacteospumosus Figure 1 & Bouché, 1969, is an earthworm within a monospecific genus and presents a very restricted distribution area, known only from a few localities covering a range of around 24 km in the region of Ariège, southern France ( Marchán et al., 2018a). Initially placed in the family Ailoscolecidae ( Bouché, 1969), it has recently been included in the family Hormogastridae ( Marchán et al., 2018b), following confirmation by phylogenomic analyses ( Novo et al., 2016). It is a family endemic to the western Mediterranean, adapted to soils prone to drought and with the capacity of aestivation in summer ( Díaz Cosín et al., 2006). Like other hormogastrids, it lacks dorsal pores and Morren glands and presents closely paired chaetae and an anterior position of the clitellum. Ailoscolex has two anterior gizzards and a multilamellar typhlosole (three lamellae). The male pore is displaced to a more posterior position in segment 22 and presents a shorter clitellum when compared to most hormogastrids (9-11 segments). It has two pairs of globular spermathecae ( Bouché, 1969).
Example image of Ailoscolex lacteospumosus.Image taken by Sylvain Gérard.
Ailoscolex is one of the basal taxa of Hormogastridae ( Novo et al., 2016) and therefore, its genome can provide valuable insights into the evolution of these Mediterranean worms. Furthermore, its extremely reduced distribution range and unique morphology highlight the importance of assessing its conservation status and implementing protection measures.
The conservation status of Ailoscolex lacteospumosus has not been formally assessed and therefore it is not listed in the IUCN Red List or any other red list. However, it presents a very limited distribution, which calls for the need to conserve this species.
Ailoscolex lacteospumosus is an endogeic earthworm species, which means that it lives in the deepest layers of the soil and builds horizontal galleries. These galleries improve water infiltration and aeration and through its feeding and burrowing, it has a significant impact on nutrient cycling, organic matter decomposition and soil structure. It therefore plays a fundamental role in terrestrial ecosystems, as described for earthworms ( Lavelle et al., 2006).
The generation of this reference resource was coordinated by the European Reference Genome Atlas (ERGA) initiative’s Biodiversity Genomics Europe (BGE) project, supporting ERGA’s aims of promoting transnational cooperation to promote advances in the application of genomics technologies to protect and restore biodiversity ( Mazzoni et al., 2023).
Materials & methods
ERGA's sequencing strategy includes Oxford Nanopore Technology (ONT) and/or Pacific Biosciences (PacBio) for long-read sequencing, along with Hi-C sequencing for chromosomal architecture, Illumina Paired-End (PE) for polishing (i.e. recommended for ONT-only assemblies), and RNA sequencing for transcriptomic profiling, to facilitate genome assembly and annotation.
Sample and sampling information
On 7th November 2023, 12 adult, hermaphroditic samples of Ailoscolex lacteospumosus were sampled by Daniel Fernández Marchán, Sylvain Gérard, and Alejandro Martínez Navarro. The species identification was confirmed via morphology and barcoding. The specimen was sampled by digging by hand in a pasture in Montesquieu-Avantès, Ariège, France. The specimen's tissues (head, anterior body and posterior body) were dissected by placing them on a glass petri dish over dry ice, kept immediately in the barcoded tubes and placed in liquid nitrogen until storage at -80. Permits are not required for sampling and sequencing of this worm, as indicated by the French Ministère de la Transition écologique, de la Biodiversité, de la Forêt, de la Mer et de la Pêche.
Vouchering information
Physical reference materials for the here sequenced specimen have been deposited in MNCN ( https://www.mncn.csic.es/es), under the accession number MNCN16.1/19304.
Frozen reference tissue material of bodywall is available from the same individual at the MNCN ( https://www.mncn.csic.es/es) under the voucher ID MNCN-ADN-151758.
Genetic information
Before sequencing, the estimated genome size, based on ancestral taxa, was 724 Mb, while the estimation based on reads kmer profiling was 479 Mb. Indirect inference of ploidy and haploid number indicate a diploid genome with a haploid number of 17 chromosomes (2n=34), however kmer profiling provides evidence that the genome is tetraploid. Estimates of genome size and karyotype for this species were retrieved from Genomes on a Tree ( Challis et al., 2023).
DNA/RNA processing
DNA was extracted from the anterior body using the Blood & Cell Culture DNA Mini Kit (QIAGEN)Kit (Qiagen) following the manufacturer’s instructions. DNA quantification was performed using a Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific), and DNA integrity was assessed using a Femtopulse system (Genomic DNA 165 Kb Kit, Agilent). DNA was stored at 4ºC until use.
RNA was extracted using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA was extracted from two different specimen body parts: anterior body and posterior body. RNA quantification was performed using the Qubit RNA BR Kit and RNA integrity was assessed using a Bioanalyzer 2100 system (RNA 6000 Pico Kit,, Agilent). RNA was pooled in a 1:1 ratio before library preparation and stored at -80ºC until use.
Library preparation and sequencing
A long-read whole genome library was prepared using the SQK-LSK114 kit and sequenced across two R10.4.1 Flow Cells on a PromethION P24 A series instrument (Oxford Nanopore Technologies). For short-read whole genome sequencing (WGS), a library was prepared using the KAPA Hyper Prep Kit (Roche). A Hi-C library preparation, using the head, was conducted with the Dovetail Omni-C Kit (Cantata Bio) and further processed with the KAPA Hyper Prep Kit for Illumina sequencing (Roche). The RNA library, generated from the pooled samples was prepared with the KAPA mRNA Hyper Prep Kit (Roche). All the short-read libraries were sequenced on the Illumina NovaSeq 6000 instrument (2x150bp). In total, 124x Oxford Nanopore, 100x Illumina WGS shotgun, and 160x HiC data were sequenced to generate the assembly.
Genome assembly methods
The genome was assembled using the CNAG CLAWS pipeline v2.2.0 ( Gomez-Garrido, 2024). Briefly, reads were preprocessed for quality and length using Trim Galore v0.6.7 and Filtlong v0.2.1 with parameters --min_length 1000 --min_mean_q 80 -t 61000000000, resulting in a read of N50=19.5 kb and median quality=Q20.6. K-mer analysis with Smudgeplot v0.2.5 ( Ranallo-Benavidez et al., 2020) proposed tetraploidy ( Figure 2), thus initial contigs were assembled using HiFiasm v0.24.0 with parameters –ont –n-hap 4 and the primary assembly was chosen for further processing. This was followed by removal of retained haplotigs using purge-dups v1.2.6 and scaffolding with YaHS v1.2a. Finally, assembled scaffolds were curated via manual inspection using Pretext v0.2.5 with the Rapid Curation Toolkit ( https://gitlab.com/wtsi-grit/rapid-curation) to remove any false joins and incorporate any sequences not automatically scaffolded into their respective locations in the chromosomal pseudomolecules (or super-scaffolds). In total 51 haplotigs were removed by purge_dups, while manual curation removed two additional haplotigs from scaffolds and two from the unplaced sequences. Summary analysis of the released assembly was performed using the ERGA-BGE Genome Report ASM Galaxy workflow ( De Panis, 2024a), incorporating tools such as BUSCO v5.5 and Merqury v1.3.
Smudgeplot ploidy estimation.Kmer-based estimation of sample ploidy as calculated by Smudgeplot. The histograms on the x- and y-axes show the normalised ratio of pair-wise kmers and total pair-wise kmer counts, respectively. “Smudges” show increased co-localisation of the two counts, and are used to estimate ploidy.
Genome annotation methods
A gene set was generated using the Ensembl Gene Annotation system ( Aken et al., 2016), primarily by aligning publicly available short-read RNA-seq data from BioSamples SAMEA117792403 and SAMEA3303732 to the genome. Gaps in the annotation were filled via protein-to-genome alignments of a select set of clade-specific proteins from UniProt ( The UniProt Consortium, 2019), which had experimental evidence at the protein or transcript level. At each locus, data were aggregated and consolidated, prioritising models derived from RNA-seq data, resulting in a final set of gene models and associated non-redundant transcript sets. To distinguish true isoforms from fragments, the likelihood of each open reading frame (ORF) was evaluated against known metazoan proteins. Low-quality transcript models, such as those showing evidence of fragmented ORFs, were removed. In cases where RNA-seq data were fragmented or absent, homology data were prioritised, favouring longer transcripts with strong intron support from short-read data. The resulting gene models were classified into two categories: protein-coding, and long non-coding. Models that did not overlap protein-coding genes, and were constructed from transcriptomic data were considered potential lncRNAs. Potential lncRNAs were further filtered to remove single-exon loci due to their unreliability. Putative miRNAs were predicted by performing a BLAST search of miRBase ( Kozomara et al., 2019) against the genome, followed by RNAfold analysis ( Gruber et al., 2008). Other small non-coding loci were identified by scanning the genome with Rfam ( Kalvari et al., 2018) and passing the results through Infernal ( Nawrocki & Eddy, 2013). Summary analysis of the released annotation was performed using the ERGA-BGE Genome Report ANNOT Galaxy workflow ( De Panis, 2024b), incorporating tools such as AGAT v1.2, OMArk v0.3, and others (see reference for the full list of tools).
Results
Genome assembly
The genome assembly has a total length of 493,041,447 bp in 24 scaffolds ( Figure 3 and Figure 4), with a GC content of 42.48%. It features a contig N50 of 15,094,000 bp (L50=10) and a scaffold N50 of 29,876,410 bp (L50=7). There are 22 gaps, totaling 4.4 kb in cumulative size. The single-copy gene content analysis using the metazoa_odb10 database with BUSCO resulted in 97.3% completeness (93.5% single and 3.8% duplicated). 56.93% of reads k-mers were present in the assembly and the assembly has a base accuracy Quality Value (QV) of 53.9 as calculated by Merqury.
Snail plot summary of assembly statistics.The main plot is divided into 1,000 size-ordered bins around the circumference, with each bin representing 0.1% of the 493,041,447 bp assembly. The distribution of sequence lengths is shown in dark grey, with the plot radius scaled to the longest sequence present in the assembly (52,692,838 bp, shown in red). Orange and pale-orange arcs show the scaffold N50 and N90 sequence lengths (29,876,410 and 19,297,223 bp), respectively. The pale grey spiral shows the cumulative sequence count on a log-scale, with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT, and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated, and missing BUSCO genes found in the assembled genome from the metazoa database (odb10) is shown on the top right.
Hi-C contact map showing spatial interactions between regions of the genome.The diagonal corresponds to intra-chromosomal contacts, depicting chromosome boundaries. The frequency of contacts is shown on a logarithmic heatmap scale. Hi-C matrix bins were merged into a 125 kb bin size for plotting.
Genome annotation
The genome annotation consists of 23,944 protein-coding genes with an associated 40,974 transcripts, in addition to 1,237 non-coding RNA genes of various types ( Table 1). Using the longest isoform per transcript, the single-copy gene content analysis using the metazoa_odb10 database with BUSCO resulted in 95.6% completeness. Using the OMAmer Lophotrochozoa database for OMArk resulted in 95.78% completeness and 61.74% consistency ( Table 2).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Aguilera L Gut M Ferreira FC : Ailoscolex lacteospumosus genome sequencing and assembly. European Nucleotide Archive.Project: PRJEB 77800 [Dataset],2024.
- 2Aken BL Ayling S Barrell D : The Ensembl gene annotation system. Database (Oxford). 2016;2016: baw 093. 10.1093/database/baw 093 27337980 PMC 4919035 · doi ↗ · pubmed ↗
- 3BouchéMB : Ailoscolex lacteospumosus, n. gen., n. sp., un ver de terre aux caractères morphologiques et biologiques remarquables (Oligochaeta, Ailoscolecidae, nov. fam.). Rev Écol Biol Sol. 1969;6(4):525–531. Reference Source
- 4Challis R Kumar S Sotero-Caio C : Genomes on a Tree (Goa T): a versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic Tree of Life [version 1; peer review: 2 approved]. Wellcome Open Res. 2023;8:24. 10.12688/wellcomeopenres.18658.1 36864925 PMC 9971660 · doi ↗ · pubmed ↗
- 5De Panis D : ERGA-BGE genome report ASM analyses (one-asm WGS Illumina PE + Hi C). Workflow Hub , 2024 a. 10.48546/WORKFLOWHUB.WORKFLOW.1103.2 · doi ↗
- 6De Panis D : ERGA-BGE genome report ANNOT analyses. Workflow Hub, 2024 b. 10.48546/WORKFLOWHUB.WORKFLOW.1096.1 · doi ↗
- 7Díaz Cosín DJ Ruiz MP Ramajo M : Is the aestivation of the earthworm Hormogaster elisae a paradiapause? Invertebr Biol. 2006;125(3):250–255. 10.1111/j.1744-7410.2006.00057.x · doi ↗
- 8Gomez-Garrido J : CLAWS (CNAG's long-read assembly workflow in Snakemake). Workflow Hub, 2024. 10.48546/WORKFLOWHUB.WORKFLOW.567.2 · doi ↗