Rare Germline Variants in CDKN2A‐Negative Children and Adolescents With Cutaneous Melanoma
Peter A. Johansson, Jane M. Palmer, Linh T. Bui‐Raborn, Mai Xu, Kristine M. Jones, Herbert Higson, Jia Liu, Kelly M. Brooks, Antonia L. Pritchard, Nicholas K. Hayward, Kevin M. Brown

TL;DR
This study finds that children and adolescents with melanoma have rare genetic mutations in genes related to telomeres, pigmentation, and DNA repair, but fewer MC1R R-alleles compared to adults.
Contribution
The study identifies rare germline variants in CDKN2A-negative early-onset melanoma patients, emphasizing the role of telomere biology and pigmentation genes.
Findings
Potentially pathogenic variants in shelterin complex genes were found in 3% of early-onset melanoma cases.
Eleven individuals (7%) had pathogenic variants in pigmentation genes linked to albinism.
Two patients carried truncating MBD4 variants, suggesting a role in early-onset melanoma.
Abstract
Cutaneous melanoma is a complex disease influenced by both environmental and genetic factors. Inherited susceptibility plays a significant role, involving a combination of high‐, intermediate‐ and low‐penetrance genes. Melanoma in children and adolescents has been speculated to have a stronger genetic component due to the early onset. This study investigates germline variants in early‐onset melanoma through exome sequencing of 154 patients in Australia diagnosed with cutaneous melanoma before the age of 20. Potentially pathogenic variants in shelterin complex genes were identified in 3% of the cases, consistent with a role for telomere dysregulation in early‐onset melanoma. MC1R R‐alleles, associated with red hair, fair skin and increased melanoma risk, were less frequent than in adult cases (0.46 vs. 0.64). Pathogenic germline variants in pigmentation genes linked to albinism were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
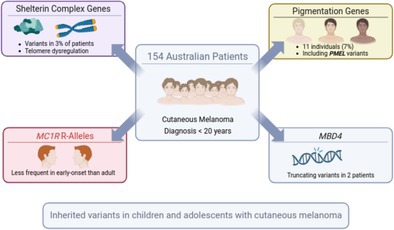 Figure 1
Figure 1- —National Health and Medical Research Council10.13039/501100000925
- —National Cancer Institute10.13039/100000054
- —Buckoff Melanoma
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCutaneous Melanoma Detection and Management · Melanoma and MAPK Pathways · melanin and skin pigmentation
Cutaneous melanoma (CM) is a complex disease influenced by both environmental and genetic factors. Inherited susceptibility plays a significant role, with rare high‐penetrance variants conferring a substantial increase in risk. The most well‐established of the susceptibility genes is in the tumour suppressor CDKN2A, which encodes the p16^INK4A^ and p14^ARF^ proteins involved in cell cycle regulation (Hussussian et al. 1994). Mutations in CDK4, another cell cycle regulator, are also strongly associated with familial CM, though they are much rarer (Zuo et al. 1996). More recently, genes involved in telomere maintenance, particularly components of the shelterin complex such as POT1, TERF2IP and ACD, as well as mutations to the promoter of the telomerase gene (TERT) itself (Horn et al. 2013), have been implicated in CM‐prone families (Aoude, Pritchard, et al. 2015; He et al. 2020; Jensen et al. 2023; Robles‐Espinoza et al. 2014; Shi et al. 2014). In addition, germline mutations in BAP1 have been linked to a tumour predisposition syndrome that includes cutaneous and uveal melanoma (Abdel‐Rahman et al. 2011; Harbour et al. 2010; Wadt et al. 2012; Walpole et al. 2018). Beyond these high‐penetrance genes, moderate‐penetrance variants also contribute to CM susceptibility. One of the most well‐characterised is MC1R, with several common variants associated with red hair, fair skin and CM risk (Landi et al. 2020; Palmer et al. 2000; Valverde et al. 1995). Other examples include DNA repair genes such as ATM (Dalmasso et al. 2021), and the MITF p.Glu318Lys variant, which confers intermediate risk by affecting the transcription of melanocyte genes (Bertolotto et al. 2011; Yokoyama et al. 2011). Genes involved in oculocutaneous albinism, including TYR, OCA2, TYRP1 and SLC45A2, have also been linked with intermediate CM risk (Landi et al. 2020; Nathan et al. 2019).
Recently, we analysed the exome from 50 paediatric CM patients (aged younger than 15 years at time of onset of CM) to investigate the contribution of rare germline variants to early‐onset CM (Johansson et al. 2023). Here, we complement that prior work by conducting a study of 154 patients diagnosed with CM in Australia before the age of 20, using exome sequencing to assess both known and candidate CM susceptibility genes (Table S1). Patients from the previous cohort were excluded from this study. With the goal of finding novel or rarer predisposing variant, childhood/adolescent cases who had previously screened for CDKN2A mutations and were found to carry a known germline pathogenic variant were excluded from this study, as were cases who belonged to families known to carry a CDKN2A mutation, irrespective of whether the individual had been sequenced for that gene. This exclusion may lead to an underestimation of the overall burden of inherited risk contributing to early‐onset CM.
Blood or saliva samples were obtained with informed written consent from the donor or their parent/guardian following approval from the Human Research Ethics Committee of QIMR Berghofer. Blood DNA was extracted using standard salting‐out methods and saliva DNA was collected and extracted using Oragene DNA kits (OG‐500, DNA Genotek, Ottawa, Canada) according to the manufacturer's instructions. Whole‐exome sequencing was performed by the NCI Division of Cancer Research Cancer Genomics Research Laboratory (using the KAPA HyperExome kit; sequencing on an Illumina NovaSeq 6000) (Frederick, MD, USA) or Macrogen (using Illumina SureSelect V7‐Post) (Seoul, South Korea). Sequence reads were mapped to the HumanG1Kv37 assembly using BWA (Li and Durbin 2010), sorted with samtools (Li et al. 2009), and duplicate reads were marked with Picard. Alignments were fine‐tuned by realigning against common variants with GATK IndelRealigner, base qualities calibrated with GATK BaseRecalibrator, variants were identified using GATK HaplotypeCaller (McKenna et al. 2010), validated with an in‐house tool, and annotated using the Variant Effector Predictor (VEP) (McLaren et al. 2016), gnomAD version 4.1 (Chen et al. 2024), AlphaMissense (Cheng et al. 2023), ClinVar categories (downloaded 8 December 2024) and PhyloP scores (Pollard et al. 2010) retrieved through the UCSC Genome Browser (Perez et al. 2025). To identify high‐quality variants we required a phred‐scaled likelihood for the wildtype to be at least 70 and we removed variants in segmentally duplicated regions (Bailey et al. 2002), variants showing mapping quality bias (MQRank > 4), or position bias (ReadPosRankSum > 4) between variant and wildtype reads. Non‐synonymous MC1R variants were categorised into R (associated with red hair) and r alleles as described previously (Robles‐Espinoza et al. 2016). To identify pathogenic variants, we focused on those with a gnomAD Non‐Finnish European allele frequency less than 0.005 and classified as high‐ or moderate‐impact by VEP, that is, variants predicted to truncate the protein or alter the amino acid sequence. Variants classified as benign or likely benign in ClinVar and missense variants not predicted to be pathogenic by AlphaMissense were excluded, unless classified as pathogenic or likely pathogenic in ClinVar.
The 154 patients were recruited from 1987 through 2023 and the median age at diagnosis was 17 years (range 5–19). We first assessed well‐established high‐penetrance melanoma susceptibility genes (Table S2). Five individuals (3%) carried variants meeting the filtering criteria described above in genes encoding components of the shelterin complex. These included two protein‐truncating variants, POT1 p.(Gln94ArgfsTer13) and TERF2IP p.(Phe21SerfsTer2), and three missense variants predicted to be pathogenic by AlphaMissense: POT1 p.(Lys33Glu), TERF2IP p.(Gly60Arg) and TERF1 p.(Cys126Arg) (Table 1). The missense POT1 p.(Lys33Glu) lies within the OB1 domain and is specifically known to bind to the telomeric single‐stranded DNA. Its classification as pathogenic by AlphaMissense, along with the saturation genome editing data, indicating a depletion of function when present (Martin et al. 2025) suggests a significant structural and functional impact on the protein, further supported by complete conservation of the affected amino acid across 100 vertebrate species (PhyloP score 5.4). The bases altered by the TERF2IP (p.Gly60) and TERF1 (p.Cys126) variants are likewise highly conserved (PhyloP score 5.1 and 4.8, respectively), which, together with their classification as pathogenic by AlphaMissense, support a potential impact on protein function. Pathogenic TERF1 variants have previously been identified in a large screen of patients with sarcoma, including one individual with familial melanoma (Ballinger et al. 2023). We did not observe potentially pathogenic variants in BAP1, CDK4, ACD or TINF2, nor did we detect any pathogenic TERT promoter mutations.
We next assessed moderate‐penetrance susceptibility genes, including MITF p.Glu318Lys, ATM and well‐established albinism and pigmentation genes. The known MITF p.Glu318Lys (Yokoyama et al. 2011) variant was identified in two individuals (1.3%), a frequency that lies between that reported in the general population (0.71%, Fisher's exact test, p = 0.3) and in adult CM patients (2.1%; Fisher's exact test, p = 0.74) (Potrony et al. 2016). The cohort size is, however, insufficient to draw statistically meaningful conclusions. We did not identify other potentially pathogenic MITF variants. Four variants were found in ATM, including a frameshifting variant p.(Ser1905IlefsTer25), splice‐site variant c.7630‐1G>A, and two missense variants p.(Pro292Leu) and p.(Tyr1442His). In terms of pigmentation genes, on average patients carried 0.46 MC1R R alleles. This is significantly lower than the 0.64 reported previously in a cohort of 303 adult CM patients studied as part of the Australian Melanoma Genome Project (Fisher's exact test, p = 0.007) and comparable to that observed in uveal melanoma (UM) patients in the same study (0.48) (Newell et al. 2022). As there is no significant association between MC1R genotypes and UM risk (Hearle et al. 2003; Mies et al. 2025) these data suggest that while R alleles are linked to an increased risk of CM (Bishop et al. 2009; Landi et al. 2020; Raimondi et al. 2008), they are not a major risk factor for childhood/adolescent CM. In our previous study of childhood CM cases (Johansson et al. 2023), the frequency of R alleles was not significantly lower than in adult cases (mean 0.56, 95% confidence interval (CI) 0.39–0.76), possibly due to the limited sample size and wide confidence interval. When combining the two childhood/adolescent cohorts the R allele frequency was 0.49 (95% CI 0.40–0.59), still significantly lower than in the Australian adult CM cases (odds ratio (OR) 0.69, Fisher's exact test, p = 0.014) (Newell et al. 2022). In comparison, in a large multicentre study of 233 young cases (most cases from Italy, the Netherlands, Spain, France and Sweden), R alleles were identified as a significant risk factor in both childhood/adolescent and adult cases, and there was no substantial difference in R allele frequency between the two groups (Pellegrini et al. 2019). The reason for this difference is not clear. It may reflect a variation in the prevalence of R alleles across different populations, as the majority of Australian patients are of British ancestry, whereas the multicentre study included a more diverse sample with greater representation from Southern Europe. Higher levels of ultraviolet radiation (UVR) in Australia than in Europe may also contribute to the difference, as well as differences in sun protection behaviour between different countries, age groups and skin type. Epidemiological studies are needed to address these possibilities.
Additionally, eight pathogenic variants were found in oculocutaneous albinism (OCA) genes, affecting OCA2 (n = 3), TYR (n = 3) and TYRP1 (n = 2) (Table 1). We also identified three individuals with germline protein‐truncating variants (PTVs) in PMEL, a gene encoding a key structural component of the melanosome, which is essential for melanin synthesis, storage and transport. PMEL has previously been linked to cancer through recurrent somatic mutations (Zhang et al. 2015). The mouse homolog of PMEL is associated with a coat colour phenotype (silver) (Kwon et al. 1995), and biallelic loss of PMEL has recently been linked to oculocutaneous albinism (AlAbdi et al. 2023). These findings suggest that monoallelic loss of PMEL contributes to susceptibility to CM, analogous to the role of other OCA‐associated genes such as TYR, OCA2, TYRP1 and SLC45A2, in which biallelic loss causes oculocutaneous albinism and heterozygous variants have been associated with increased CM risk (Landi et al. 2020; Nathan et al. 2019).
Four individuals carried variants in genes previously linked to non‐sun‐exposed melanoma subtypes. Two individuals harboured PTVs in MBD4 p.(Glu314ArgfsTer13) and p.(Tyr539Ter), a gene associated with an increased risk of uveal melanoma (Derrien et al. 2021; Villy et al. 2024). We also previously reported a single case of an MBD4 PTV in a cohort of 50 paediatric CM patients (Johansson et al. 2023), bringing the total to three carriers (1.5%), suggesting that MBD4 may predispose to early‐onset CM. Another individual carried a missense variant in PLCB4 (p.Ser342Leu), predicted to be pathogenic by AlphaMissense. However, this variant differs from the well‐characterised gain‐of‐function PLCB4 mutation (p.Asp630), which is often somatically acquired in uveal melanoma tumours and activates the Gq/11‐dependent signalling pathway (Johansson et al. 2016). Finally, one individual carried a missense variant in WRN, p.(Lys483Asn), which is predicted to be pathogenic by AlphaMissense. WRN is primarily associated with Werner syndrome but has also been linked to acral melanoma (Goto et al. 1996; Newell et al. 2022). The carrier reported here had no record of acral melanoma.
Lastly, we examined genes linked with other cancers (Table S1), restricting our analysis to rare variants with a gnomAD Non‐Finnish European allele frequency less than 5 × 10^−4^ (Table S3). One individual carried a TP53 p.(Lys120Asn) variant, a missense alteration predicted to be pathogenic by AlphaMissense. The variant is extremely rare in the germline, having not been observed in a dataset of 800,000 individuals (gnomAD 4.1) and is therefore absent from clinical variant databases such as ClinVar. Somatically, it has been classified as a recurrent hotspot (Chang et al. 2016), including in databases such as cBioPortal. It was also included in a systematic functional screen, in which it received a relative fitness score of −0.135, intermediate between the scores of silent variants (mean −2.50) and PTVs (mean 0.42), suggesting a potential intermediate impact on the p53 function (Kotler et al. 2018).
Four individuals collectively carried variants in POLE p.(Cys501Ter), p.(Pro282Ser), p.(Lys284Glu), or POLD1 p.(Leu983ArgfsTer65), two genes encoding proteins involved in DNA proofreading. Both missense variants occurred in the exonuclease domain (amino acids 268–471), consistent with a likely impact on proofreading capability. Loss of function of these proteins has primarily been linked to colon cancer but has also been reported in other cancers, including cutaneous melanoma (Aoude, Heitzer, et al. 2015; Mur et al. 2020). Apart from the four variants in ATM, another 13 variants were identified in genes involved in homologous recombination repair, including five PTVs in BRIP1, BARD1, CHEK2, FANCA and FANCF. Three individuals carried pathogenic variants in XPA, XPC and ERCC1, genes involved in nucleotide excision repair. Biallelic loss of these genes causes xeroderma pigmentosum, a rare recessive disorder characterised by extreme sensitivity to UV radiation. We recently reported two patients affected by both uveal and cutaneous melanoma who carried pathogenic variants in XP genes (Johansson et al. 2025). The presence of these three variants in this cohort further supports the hypothesis that monoallelic loss of these genes increases UV sensitivity and elevates CM risk.
One individual carried a DNMT3A p.(Arg882Cys) variant affecting a well‐characterised hotspot for somatic mutations in myeloid leukaemias (Ley et al. 2010). This mutation is a known early event in disease development, frequently observed in clonal haematopoiesis and myeloid malignancies. In contrast, germline mutations in DNMT3A cause Tatton–Brown–Rahman syndrome, a development disorder associated with overgrowth and neurodevelopment delay (Tatton‐Brown et al. 2014). Notably, the carrier did not exhibit features of this syndrome but was diagnosed with a haematological malignancy at age 73, approximately a decade after donating blood for this study, suggesting that the DNMT3A variant had likely been acquired somatically and therefore unrelated to her early‐onset melanoma. We observed another five pathogenic variants in genes linked with haematological malignancies, including ROS1 p (Asp839Glu), JAK3 p.(Val946Leu) and three PTVs in RUNX1 (Table S3), also possibly acquired somatically.
Several individuals carried more than one potentially pathogenic variant. For example, MELA_1082 and MELA_1043 carried variants in PMEL/TP53 and TYR/TERF2IP, respectively (Table 1). Others carried combinations such as ATM/MITF, CHEK2/POLD1, POT1/POLE, ATM/TERF1, and OCA2/FANCF (Tables S2 and S3). These findings highlight several observations: first, they support the notion that many of these variants have intermediate penetrance, consistent with previous suggestions for OCA genes (Nathan et al. 2019) and population‐based estimates of POT1 variant penetrance (OR = 2.11) (Simonin‐Wilmer et al. 2022). Second, there is a trend where variants in pigmentation genes such as PMEL, MITF and OCA2 co‐occurred with variants in DNA repair genes such as TP53, ATM, XPA, POLE and FANCF. This likely reflects the multilayered protection melanocytes rely on against mutagenic UVR with melanin acting as a physical shield, followed by repair mechanisms that correct UVR‐induced DNA damage. Similarly, the co‐occurrence of variants in shelterin genes (TERF2IP, POT1 and TERF1) with pigmentation or DNA repair genes reminds us that while telomere maintenance is required for limitless replication, it is not sufficient for tumorigenesis. Aside from germline variants, a melanocyte must acquire multiple cancer‐enabling mutations somatically to become malignant (Hanahan and Weinberg 2011). In contrast to adult cancer, where such acquired changes may be accumulated over extended periods of time, in childhood/adolescent cancers there may be greater reliance on inherited germline variants.
In conclusion, five individuals (3%) carried pathogenic variants in POT1, TERF1 or TERF2IP, supporting a significant role for shelterin complex genes in early‐onset CM. A lower‐than‐expected frequency of MC1R R‐alleles, typically associated with fair skin and melanoma risk, was observed. Instead, 11 individuals (7%) carried pathogenic germline variants in pigmentation genes linked to oculocutaneous albinism, including three with protein‐truncating variants in PMEL, reinforcing the role of pigmentation genes in CM predisposition. Truncating variants in MBD4 were identified in two individuals; together with a previously reported paediatric case, these findings are consistent with a potential role for MBD4 in early‐onset CM. The high frequency of rare variants associated with high or intermediate melanoma risk underscores the importance of incorporating such variants into genetic risk assessments. As our understanding of melanoma genetics evolves, comprehensive risk profiles will be crucial for interpreting genetic tests and guiding long‐term cancer surveillance in both early‐onset CM patients and their at‐risk relatives.
Author Contributions
Conceptualisation and funding acquisition: N.K.H.; data curation: P.A.J., J.M.P., L.T.B.‐R., A.L.P.; formal analysis, software and writing – original draft preparation: P.A.J.; methodology: P.A.J., A.L.P.; investigation: L.T.B.‐R., M.X., K.M.J., H.H., J.L., K.M.B., resources: J.M.P., N.K.H., Kev.M.B, writing – review and editing: P.A.J., J.M.P., L.T.B.‐R., M.X., K.M.J., Kel.M.B., A.L.P., N.K.H. and Kev.M.B.
Funding
This work was supported by the National Health and Medical Research Council (APP1195581), National Cancer Institute, Buck Off Melanoma.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1: List of cancer genes. Table S2: Variants identified in cutaneous melanoma susceptibility genes. Table S3: Variants identified in other cancer genes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdel‐Rahman, M. H. , R. Pilarski , C. M. Cebulla , et al. 2011. “Germline BAP 1 Mutation Predisposes to Uveal Melanoma, Lung Adenocarcinoma, Meningioma, and Other Cancers.” Journal of Medical Genetics 48, no. 12: 856–859. 10.1136/jmedgenet-2011-100156.21941004 PMC 3825099 · doi ↗ · pubmed ↗
- 2Al Abdi, L. , M. Alshammari , R. Helaby , A. O. Khan , and F. S. Alkuraya . 2023. “PMEL Is Mutated in Oculocutaneous Albinism.” Human Genetics 142, no. 1: 139–144. 10.1007/s 00439-022-02489-y.36166100 · doi ↗ · pubmed ↗
- 3Aoude, L. G. , E. Heitzer , P. Johansson , et al. 2015. “POLE Mutations in Families Predisposed to Cutaneous Melanoma.” Familial Cancer 14, no. 4: 621–628. 10.1007/s 10689-015-9826-8.26251183 · doi ↗ · pubmed ↗
- 4Aoude, L. G. , A. L. Pritchard , C. D. Robles‐Espinoza , et al. 2015. “Nonsense Mutations in the Shelterin Complex Genes ACD and TERF 2IP in Familial Melanoma.” Journal of the National Cancer Institute 107, no. 2: dju 480. 10.1093/jnci/dju 408.PMC 433478725505254 · doi ↗ · pubmed ↗
- 5Bailey, J. A. , Z. Gu , R. A. Clark , et al. 2002. “Recent Segmental Duplications in the Human Genome.” Science 297, no. 5583: 1003–1007. 10.1126/science.1072047.12169732 · doi ↗ · pubmed ↗
- 6Ballinger, M. L. , S. Pattnaik , P. A. Mundra , et al. 2023. “Heritable Defects in Telomere and Mitotic Function Selectively Predispose to Sarcomas.” Science 379, no. 6629: 253–260. 10.1126/science.abj 4784.36656928 PMC 12147039 · doi ↗ · pubmed ↗
- 7Bertolotto, C. , F. Lesueur , S. Giuliano , et al. 2011. “A SUM Oylation‐Defective MITF Germline Mutation Predisposes to Melanoma and Renal Carcinoma.” Nature 480, no. 7375: 94–98. 10.1038/nature 10539.22012259 · doi ↗ · pubmed ↗
- 8Bishop, D. T. , F. Demenais , M. M. Iles , et al. 2009. “Genome‐Wide Association Study Identifies Three Loci Associated With Melanoma Risk.” Nature Genetics 41, no. 8: 920–925. 10.1038/ng.411.19578364 PMC 2741419 · doi ↗ · pubmed ↗