The role of epigenetic modifications in hematological cancers
Jovana Ilic, Anna Bold, Stefan Knop

TL;DR
This paper reviews how changes in epigenetic regulation contribute to the development and progression of blood cancers like leukemia and lymphoma.
Contribution
The paper provides a comprehensive review of epigenetic alterations in hematological cancers and their impact on oncogenesis.
Findings
Mutations in enzymes regulating DNA methylation and histone modifications are linked to chromatin changes in blood cancers.
Altered epigenetic mediators influence transcriptional profiles, promoting cancer progression and drug resistance.
Key epigenetic changes are identified in leukemias, lymphomas, and multiple myeloma.
Abstract
Epigenetic regulation of gene expression entails DNA methylation and histone modifications, which orchestrate chromatin structure and transcriptional activity. Aberrant regulation of these mechanisms contributes to the development and progression of hematological cancers. Mutations in enzymes mediating DNA methylation and histone modification patterns reshape chromatin structure, influencing transcriptional profiles, thus promoting oncogenesis, clonal evolution and drug resistance. This review focuses on significant alterations in DNA methylation patterns, mutations and changes in expression of histone modifying enzymes and chromatin remodeling complexes described in leukemias, lymphomas and multiple myeloma. We summarize the most prominent changes in epigenetic mediators and their impact on transcriptional activity and oncogenesis.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
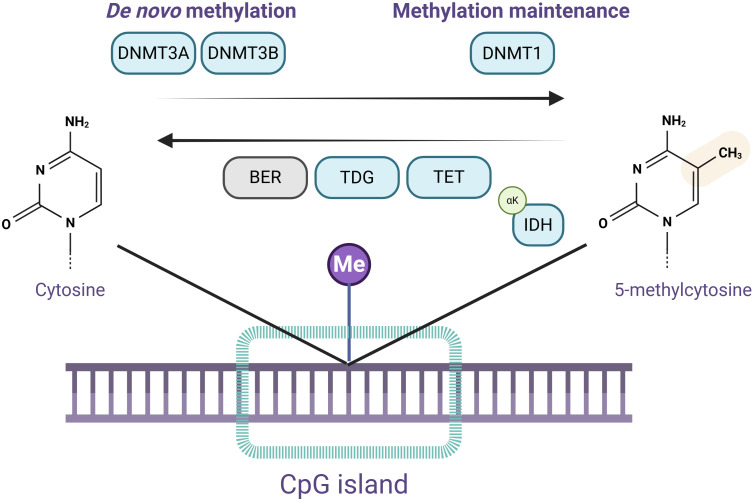 Figure 1
Figure 1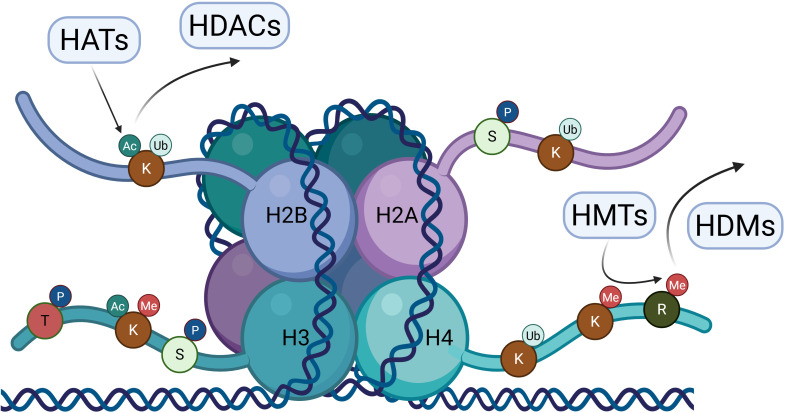 Figure 2
Figure 2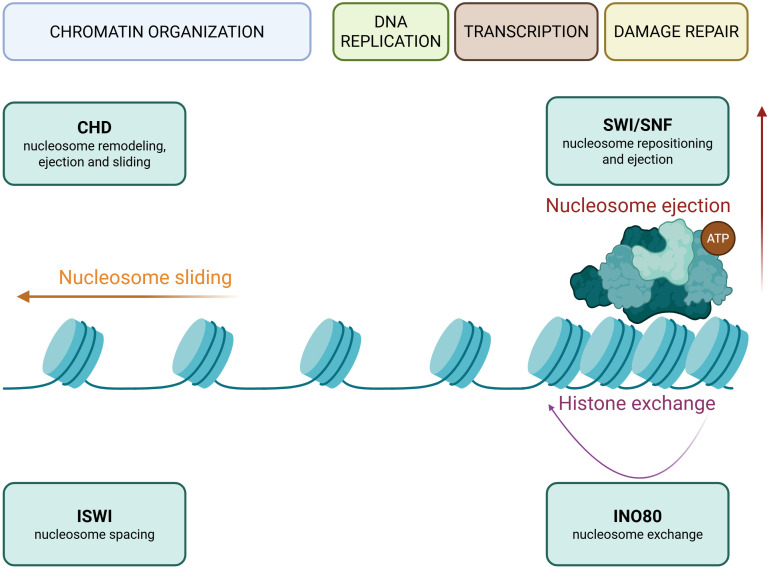 Figure 3
Figure 3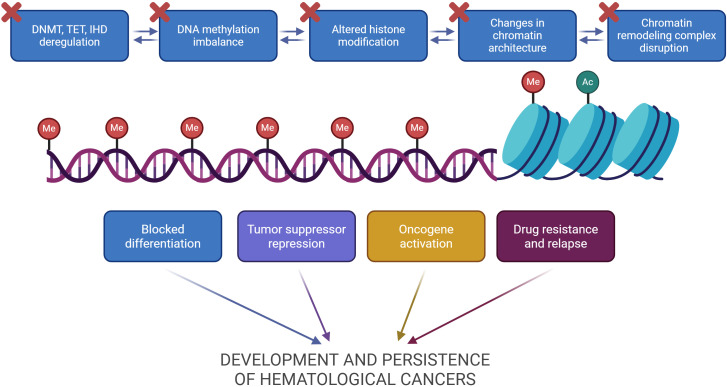 Figure 4
Figure 4| Disease | AML | ALL (B/T) | MDS |
|---|---|---|---|
| DNA (de)methylation enzymes | High frequency of mutations in | Less frequent | High incidence of |
| DNA methylation pattern | Global hypomethylation and promoter hypermethylation of tumor suppressors, used as a prognostic marker ( | Hypomethylation of tumor suppressor genes ( | The progression of MDS to AML is correlated with DNA methylation of tumor suppressor genes ( |
| Histone modifications | Overexpression of | ||
| Chromatin remodeling complexes | High expression of | Less frequent than AML, suggesting a developmental role of these lesions. | |
| Functional consequences | Blocked differentiation and persistence of drug-resistant clones ( | Pathological progenitor self-renewal and impaired differentiation ( | Increased self-renewal and survival of drug-resistant clones ( |
| Disease | CLL | Multiple myeloma (MM) | Lymphomas (B/T, FL, DLBCL, AITL) |
| DNA (de)methylation enzymes | No distinct DNMT/TET/IDH mutations have been identified. | ||
| DNA methylation pattern | Methylation patterns do not correlate with therapy response ( | Global hypomethylation with focal hypermethylation of tumor suppressors and differentiation regulators; epigenetic remodeling drives MGUS to MM ( | DNA methylation as a prognostic marker in B-cell lymphoma ( |
| Histone modifications | Upregulated | Oncogenic increase of EHZ2 activity across lymphomas ( | |
| Chromatin remodeling complexes | Very high frequency of | ||
| Functional consequences | DNA damage repair and lymphocyte differentiation gene expression dysregulation ( | Increased self-renewal, inhibition of apoptosis, repression of tumor suppressors and B-cell enhancers, oncogene overactivation ( | Disrupted normal B-cell development, inability to exit the GC (FL and DLBCL) ( |
- —Stifterverband10.13039/501100008384
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · Acute Myeloid Leukemia Research · Protein Degradation and Inhibitors
Introduction
1
Epigenetics is a broad term that encompasses several different cellular mechanisms that alter gene expression independently of the DNA sequence. Epigenetic regulation of gene expression facilitated the evolution of multicellular organisms, enabling a dynamic control of cellular processes and differentiation necessary for diverse tissue formation and complexity (1). Epigenetic processes include DNA methylation, histone modifications, and chromatin remodeling. These mechanisms allow swift cellular responses to ever-changing cellular signals through alterations in DNA conformation and its transcriptional availability (2). In addition, regulatory non-coding RNAs (ncRNAs) are non-protein coding transcripts which regulate transcription, translation, splicing, and chromatin remodeling, influencing all cellular functions. Even though ncRNAs are closely involved in epigenetic processes, they are not an epigenetic mechanism per se. Given the vast diversity of their sizes, roles and mechanisms of action, they could be considered a distinct entity and will therefore not be discussed in this review (3).This review will focus on classical epigenetic mechanisms (DNA methylation, histone modifications and chromatin remodeling complexes) to maintain a defined scope.
Hematological cancers originate from cells generated via hematopoiesis and include leukemias, lymphomas, and multiple myeloma (4). Hematopoiesis is a closely regulated process of generating new components of circulating blood cells and the immune system from a common hematopoietic stem cell progenitor (5). To date, many epigenetic factors have been identified to play a role in this complex, multi-stage differentiation process. Cancer cells from the same individuals differ in their phenotype and functionality, generating a heterogeneous population, and are thus difficult to therapeutically target at once. Therefore, it has become clear that epigenetic involvement plays a crucial role in the development, maintenance, and resistance of malignancies. In the context of hematological malignancies, aberrant DNA methylation and mutations in histone modification and chromatin remodeling enzymes are observed frequently (6). Unraveling the epigenetic patterns of these cancers is essential for their deeper understanding and improved clinical outcomes.
DNA methylation in hematological malignancies
2
DNA methylation includes the addition of a methyl group to cytosine, within the cytosine-guanine dinucleotides (CpG). It is conveyed by DNA methyltransferase (DNMT) enzymes and it is widely spread out over the whole genome (7). The DNA methylation pattern at cytosine residues within CpG sequences is established during early blood cell development and inherited through cell divisions. Many transgenic regions in the genome, including transposons, are regulated via methylation. DNMT1 is mainly involved in the methylation maintenance during replication, while DNMT3, especially DNMT3A, and DNMT3B, are responsible for the de novo methylation in the genome (8). Widespread genomic hypomethylation is often observed in cancer, coinciding with the activity of mobile genetic elements and genomic instability, contributing to higher mutagenicity. In addition, many tumor suppressor genes and microRNAs (miRNAs) are silenced by promoter hypermethylation in cancer cells (9, 10). Both DNMT1 and DNMT3 are strongly linked to hematological malignancies. DNMT1 is frequently overexpressed in cancer cells and seems to promote tumor development, whereas DNMT3A is regarded as tumor suppressive, often downregulated and mutated in hematological cancers. DNMT3B exhibits a dual role in hematological malignancies, with data confirming both its oncogenic and tumor suppressive functions (11, 12). On the other hand, members of the ten-eleven translocation methylcytosine dioxygenase (TET) family of enzymes are involved in DNA demethylation and are often found to be mutated in hematological malignancies as well (13). TET enzymes oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), an intermediate step in the DNA demethylation process. TET generated intermediates are recognized and excised by thymine-DNA glycosylase (TDG), and cytosine is restored through the base excision repair (BER) mechanism (14). A prominent member of the TET family, TET2, is highly expressed in hematopoietic stem and progenitor cells and regulates self-renewal and differentiation. This function correlates to its role in hematological cancers originating from immature progenitors (15).
An overview of DNA methylation is provided in Figure 1.
Overview of DNA methylation and demethylation pathways. Schematic representation of DNA (de)methylation pathways within the CpG islands. DNA methyltransferases DNMT3A and DNMT3B catalyze de novo methylation, forming 5-methylcytosine. DNMT1 is responsible for methylation maintenance of newly synthesized DNA strands during replication. Active DNA demethylation is carried out by Ten–Eleven Translocation (TET) enzymes, which oxidize 5-methylcytosine into intermediates recognized and excised by Thymine DNA Glycosylase (TDG). The excised nucleotide is repaired by the base excision repair (BER) machinery, restoring cytosine. Isocitrate Dehydrogenase (IDH) enzymes produce αKG, a cofactor necessary for TET activity. Image created in Biorender.
Mutations in DNA methyltransferase enzymes
2.1
To date, DNMT3A mutations have been identified in most hematological malignancies (16–20). For example, DNMT3A mutations are identified in one third of acute myeloid leukemia (AML), myeloproliferative neoplasia (MPN) and myelodysplastic syndrome (MDS) patients (21). Loss of DNMT3A function disrupts hematopoietic cell differentiation, favoring an expansion of hematopoietic stem cells which have the ability to self-renew (22). Self-renewing cells present a significant treatment hurdle, as they usually represent the drug-resistant cancer cell reservoir (23). Notably, hematopoietic stem cells from AML patients with DNMT3 mutations displayed increased treatment resistance and a potential for secondary mutations and relapse, with shortened median overall survival (16, 24). The activity of DNMT3 is correlated with tumor suppression and its mutation or downregulation is considered an early driver event in leukemia and multiple myeloma (MM) development as well. In fact, the promoter of the DNMT3A itself was hypermethylated in MM cells, causing its reduced expression (21, 25, 26). However, DNMT3 mutations were not correlated with their exact functions in these cancers and their fundamental consequences remained unknown.
DNMT1 overexpression is a frequent occurrence in other hematological cancers and leads to silencing of tumor suppressor genes due to promoter hypermethylation (27, 28). For instance, in MM, increased DNMT1 protein activity downregulates the tumor suppressor cyclin-dependent kinase inhibitor 2A (p16, CDKN2A), leading to dysregulated proliferation (29). Similarly, the increased activity of DNMT1 was observed across leukemia and related to demethylation of its promotors (30). Functionally, this led to tumor suppressor silencing via promotor hypermethylation, such as cyclin-dependent kinase inhibitor 2B (p15, CDKN2B) in AML, Src Homology region 2 domain-containing Phosphatase-1 (SHP1) in chronic myeloid leukemia (CML), and Phosphatase and Tensin Homolog (PTEN) in B-cell Acute Lymphoblastic Leukemia (B-ALL) (11, 31, 32). In highly proliferating cancer cells, where DNA replication is prominent, the overexpression of DNMT1 might initially occur as a regulatory response. Consequently, this leads to hypermethylation of cell cycle regulatory genes and promotes proliferation. DNMT1 overexpression is associated with drug resistance, poor clinical prognosis, enhanced stem-like characteristics, and increased aggressiveness. These outcomes are a consequence of a disturbed cell cycle and hypermethylation of regulatory protein promoters (11).
The activity of DNMT3B enzymes was investigated less. There are several studies confirming their contrasting roles in hematological cancers, depending on the tumor microenvironment, catalytic activity, and the experimental system applied for its investigation. For instance, loss of DNMT3B catalytic activity caused decreased methylation of the oncogenic mesenchymal–epithelial transition factor (Met) promoter in mice, leading to its upregulation. In turn, this resulted in increased Signal transducer and activator of transcription 3 (STAT3) phosphorylation and lymphomagenesis (12). In humans, the DNMT3B promoter was methylated in some cases, causing its downregulation, which led to tumor growth and elevated stemness in AML cells (33). Taken together, these studies suggest the tumor suppressive role of DNMT3B.
Nonetheless, the oncogenic activity of DNMT3B is discussed, as the levels of this enzyme were increased in T-ALL and Burkitt’s lymphoma, coinciding with increased oncogenic activity of myelocytomatosis (MYC) (34). However, the increased DNMT3B expression may occur as a consequence of a positive feedback loop, with the goal to reduce the activity of c-Myc, indicating a tumor suppressive role. This is supported by the fact that a knockdown of MYC in both T-ALL and Burkitt’s lymphoma cell lines caused a downregulation of DNMT3B (34). No conclusive data has been acquired on alternative transcript processing as a potential underlying cause of the enzyme’s different functionality in hematological cancers. However, it is indicated that a truncated isoform of this enzyme, DNMT3B7, which is catalytically inactive and commonly found in cancer cells, contributes to Myc-driven lymphomagenesis in a transgenic mouse model (35). This suggests that isoform presence, rather than total DNMT3B levels, might determine the biological outcome, but this remains unexplored in human primary samples. In addition, the oncogenic role of specific enzymes can be disease-specific, since these malignancies have their own characteristics, different origins, and a variable proteomic milieu.
Mutations in ten-eleven translocation methylcytosine dioxygenase enzymes
2.2
Members of the TET family of enzymes, such as TET1, TET2, and TET3 are generally considered tumor suppressors in hematological cancers, since their deregulation contributes to disease development (13). TET2 mutations are more frequent and are associated with myeloid malignancies, whereas TET1 mutations are found in lymphoid malignancies and are not that common overall (36–38). TET1 is often silenced epigenetically in hematological cancers, including follicular lymphoma (FL) and MM, through promoter methylation (39). In contrast, TET2 is one of the most frequently mutated genes in myeloid malignancies:10-30% in AML, 20-30% in MDS, and up to 65% in chronic myelomonocytic leukemia (CMML) cases (38, 40). These mutations often coincide with other recurrent genetic lesions, including DNMT3A and isocitrate dehydrogenase 1/2 (IDH1/2), indicating that TET2 loss cooperates with additional molecular events to drive leukemogenesis (41).
Despite their prevalence, no mutational hotspots have been identified in the TET2 gene in hematological cancers, as the mutations were distributed throughout the entire coding region (42). Additionally, these mutations were not recognized in other somatic cells, suggesting that TET2 loss of function may be a driver event in hematologic carcinogenesis (43). In AML, the loss of TET2 is a consequence of nonsense or missense mutations and frameshift indels within the catalytic domain, resulting in impaired enzymatic activity. In murine in vivo models, Tet2 loss of function resulted in elevated self-renewal of hematopoietic cells, coinciding with disturbed hematopoietic differentiation and an abnormal accumulation of immature myeloid cells (44, 45). Dysregulated differentiation directly contributes to the development of clonal hematopoiesis, which clinically manifests as increased risk for cancer progression and drug resistance, with poorer overall survival rates. In addition, TET2 loss in hematopoietic stem cells causes an increased mutational burden, observed both in mice and myeloid malignancy patients (46). This is consistent with clinical data showing how TET2-mutated clones persist after treatment and lead to clonal evolution and relapse in AML patients (47). The elevated mutational load creates an environment for additional oncogenic mutations, linking TET2 deficiency to disease development. TET2 mutations were associated with decreased levels of 5hmC in MPN, CMML, MDS, and AML patient samples (48, 49). Reduced 5hmC levels disrupt regular gene expression patterns, favoring the survival of immature, persistent clones. This leads to therapy resistance and higher relapse rates. In mice, specific Tet2 mutations were linked to extramedullary hematopoiesis, increased repopulation capacity, splenomegaly and elevated myelopoiesis, which could not have been compensated by the other two TETs (50, 51).
While TET2 has been extensively investigated in hematological malignancies, less is known about TET3. TET3 is involved in the maintenance of genomic stability in hematopoietic cells (52). In some lymphoid malignancies, the downregulation of TET3 has been observed, but its precise role in carcinogenesis is not described (36).
TET loss of function contributes to hematological cancers through many synergistic mechanisms, which are not yet fully understood. For example, loss of TET proteins changes DNMT3A localization and can paradoxically contribute to DNA hypomethylation, which promotes genomic instability and disrupts hematopoiesis (53). These changes lead to aberrant self-renewal and differentiation in hematopoietic progenitors, promoting oncogenesis. Taken together, current studies indicate a correlation between TET impairment and a more stem-like behavior of cancer cells, increasing their chances of survival and leading to drug resistance and relapse.
Mutations in isocitrate dehydrogenase enzymes
2.3
IDH enzymes are indirectly involved in the DNA demethylation process. They mediate the conversion of isocitrate to α-ketoglutarate (αKG), a cofactor of TETs. Mutations in IDH genes result in the inhibition of TET enzyme activity which disrupts normal epigenetic regulation and cell differentiation (54). Mutated IDH enzymes mediate the production of an oncometabolite, D-2-hydroxyglutarate (2-HG), which mimics αKG and impacts TET function, influencing epigenetic regulation (55).
In AML, IDH1 and IDH2 mutations were recognized in about 20% of patients. This correlated with specific (and similar) methylation profiles, with a hypermethylated DNA signature (54, 56). Despite their high prevalence, IDH1/2 mutations rarely co-occur with TET2 mutations in AML. These mutations ultimately result in TET disfunction and seem to be mutually exclusive (57).
Besides AML, IDH mutations were not reported in other hematological cancers with such frequency, except for a very rare disease, angioimmunoblastic T-cell lymphoma (AITL), where they are found in up to 45% of patients (58–60). In AILT, IDH2 mutations coincide with TET2 and DNMT3A mutations in T follicular helper cells. Acting synergistically, these mutations contribute to genomic instability and oncogenesis (61). Unlike in AML, IDH2 mutations might not be sufficient to inhibit TET activity on their own in AITL. This suggests that both TET2 and DNMT3A loss is needed to disrupt the genome organization.
Since they might follow different epigenetic patterns, different hematological cancers (especially those of different origin) should be investigated separately, with the aim of identifying shared underlying principles. Taken together, identified mutations in both DNMT, TET, and IDH gene families highlight that the balance and patterns of DNA methylation are key distinguishing factors between healthy and neoplastic cells.
Specific methylation patterns correlated with hematological malignancies
2.4
Despite extensive data collected on mutations in enzymes involved in DNA (de)methylation, their functional consequences, enzymatic activity, and clinical relevance are not always defined. DNA methylation patterns, a result of these enzymatic processes, may provide deeper insights into the development and maintenance of hematological cancers. Global hypomethylation and focal hypermethylation of DNA are well recognized in hematological malignancies.
In myeloid neoplasms (AML and MDS), specific promoters of genes involved in the inhibition of Wingless and Int-1 (WNT) and mitogen activated protein kinase (MAPK) signaling pathways were hypermethylated, compared with healthy donor CD34^+^ bone marrow cells. This resulted in the overactivation of these pathways, sustaining an immature cellular phenotype, consistent with the impaired differentiation linked to the development of these cancers (62). The progression of MDS to AML is believed to be driven by aberrant methylation of tumor suppressor genes (63). Accordingly, the identification of DNA methylation profiles, or epitypes, might support AML patient stratification as it provides insights into disease pathogenesis and could dictate therapeutic strategies (64). For example, Kelly et al. identified specific CpG island DNA methylation phenotypes in AML that correlated with disease-free survival (65). Currently, DNA methylation-based bioinformatics tools are being developed to improve diagnostic predictions and early detection in patients with AML and B-cell Non-Hodgkin Lymphoma (66, 67).
Similarly, in MM, DNA methylation patterns correlate with the stage of the disease and can be used as biomarkers (26). MM can be preceded by a non-malignant condition called monoclonal gammopathy of undetermined significance (MGUS). This transition coincides with global hypomethylation, focal hypermethylation, and altered DNMT3 expression (26, 68). Extensive hypermethylation of B-cell specific enhancers and cyclin-dependent kinase inhibitors, as well as Wnt antagonists leads to a less differentiated, more aggressive stem-like cancer cell phenotype (69, 70). Comparable to AML, hypermethylation of promoter regions in Wnt signaling pathway inhibitor genes led to an overactivation of Wnt signaling in both MM cell lines and primary MM samples, which correlates with a less differentiated MM phenotype (71, 72). This is another example of DNA methylation patterns correlating with stem-like traits of cancer cells, directly affecting their drug response and relapse. To that matter, demethylation of the ATP-binding cassette transporter G2 (ABCG2) promoter is treatment inducible in MM cells and causes its overexpression, increasing drug resistance (73).
In lymphoid malignancies such as ALL, certain tumor suppressor gene promoters were methylated. However, these methylation patterns were often stochastic and contributed to the heterogeneity of the disease (74, 75).
In contrast, in Chronic Lymphocytic Leukemia (CLL) patients, no specific methylation profiles were correlated with treatment response, likely due to its indolent biology (76).
Most DNA methylation alterations in hematological malignancies follow similar patterns, which ultimately lead to increased oncogenic activity and tumor suppressor inactivation. However, this conclusion might stem from research bias, as most studies focus on specific loci methylomes, particularly within the coding regions, driven by an interest in the functional consequences of the methylation. Not much research has been conducted in recent years on this topic, although additional studies and analyses with the data already available might provide new insights due to the rise in the simplicity and computational power of (bio)informatics tools.
Histone modifications in hematological malignancies
3
Histones are proteins that provide structural support to DNA. Together with histones, DNA forms nucleosomes – the basic building units of chromatin. Histone modifications are posttranslational changes of histones including phosphorylation, ubiquitination, acetylation, methylation, sumoylation, etc. (Figure 2). Methylation and acetylation are most commonly investigated in cancer and their functions are well described (77). Ubiquitination of histones occurs as a regulatory signal and seldom as a mark for protein degradation, as there is no sequential addition of this peptide (78). Specific combinations of histone modifications comprise the histone code, which determines chromatin structure and the recruitment of downstream effector proteins. The crosstalk between DNA and histone modifications directs the interactions with protein complexes involved in replication, transcription, DNA repair and chromatin structure. Thus, modification of histones is responsible for gene expression regulation and genome stability. Disruption of the balance of histone modifications is involved in tumorigenesis, as it leads to dysregulated gene expression and genome instability (77).
Histone modification landscape and major enzymes. Schematic representation of a nucleosome composed of the histone H2A, H2B, H3 and H4 octamer with DNA wrapped around it. Amino acids located on the N-terminal histone tails such as lysine (K), serine (S), threonine (T) and arginine (R) are modified post-translationally. The most common histone modifications, such as methylation (Me), acetylation (Ac), phosphorylation (P) and ubiquitination (Ub) are shown in the image. Histone methyltransferases (HMTs) catalyze the addition of methyl groups on lysine or arginine residues, while histone demethylases (HDMs) catalyze their removal. Histone acetyltransferases (HATs) and deacetylases (HDACs) are involved in the addition or removal of acetyl groups from lysine residues. These dynamic combinations of histone modifications regulate chromatin structure and gene expression by modulating DNA accessibility and downstream protein recruitment. Image created in Biorender.
Histone methylation in hematological malignancies
3.1
Methylation of histones is mediated by histone methyltransferase enzymes (HMTs) and histone demethylases (HDMs). It entails the addition or removal of methyl groups to the lysine or arginine residues of (mostly H3 and H4) histone proteins. Histone methylation is considered to repress transcription, but can also be involved in transcription activation, depending on the methylated histone, degree of methylation and the amino acid (position) (79, 80).
Mutations in histone methylation proteins often affect stemness and differentiation in hematological malignancies and are described extensively in cancers arising from immature progenitors, such as AML and ALL. Rearrangements of the lysine methyltransferase 2A (KMT2A) gene in cancer cells were recognized a while ago as drivers of ALL, particularly in children (81). KMT2A translocation results in aberrant fusion proteins and occurs in mixed lineage leukemia (MLL), a subtype of the disease characterized by this mutation (82). KMT2A is essential for hematopoietic stem cell development and maintenance, which explains its role in immature blood cancers (83). Mutations in this gene affect chromatin structure and gene expression regulation and are directly involved in ALL pathogenesis via H3 lysine methylation (H3K4) (82). H3K4 methylation in the promoter region facilitates the transcription of Homeobox protein Hox-A9 (HOXA9) in both ALL and AML (84, 85),. Upregulation of HOXA9 promotes pathological progenitor self-renewal and impaired differentiation, which can lead to more aggressive tumor behavior and poor clinical outcomes (86, 87). KMT2A rearrangements can also be regarded as a prognostic marker in ALL and AML. In AML, up to 10% of patients exhibit KMT2A translocations, resulting in aberrant fusion proteins, and their frequency can increase upon treatment (88). This post-therapy increase in KMT2A-mutated clones supports the idea that these mutations govern a persistent, self-renewing subpopulation capable of drug resistance and tumor repopulation.
Besides KMT2A, additional histone methylation enzymes are involved in the development and persistence of AML. For instance, dysregulation of Polycomb repressive complex 2 (PRC2) histone methylation is involved in AML stemness via the activity of the enhancer of zeste homolog 2 (EZH2) subunit (89). EZH2 methylates H3K27, a modification involved in the repression of gene expression (90). During early disease development, EZH2 activity is necessary for epigenetic repression of differentiation, positively impacting AML survival and proliferation (91). However, a recent study showed that EZH2 loss-of-function mutations led to increased chemotherapy resistance in AML cell lines. This is caused by the upregulation of EZH2 target genes involved in proliferation, apoptosis evasion, and membrane transport (92). This discrepancy largely occurs due to the context- and stage-dependent roles of the enzyme, as well as the utilization of different disease models. In particular, one study describes the importance of EZH2 in progenitor cells and the other in therapy-stressed cell lines with a different adaptation profile. In addition, different therapeutic modalities and model systems can cause an entirely distinct transcriptomic profile in the cells, which dictates the function of EZH2.
EZH2 activity is associated with other hematological cancers, such as MM, B-cell non-Hodgkin lymphoma, FL, CML, and natural killer/T-cell lymphoma (93–97). In B-cell lineage malignancies, EZH2 activity seems to be oncogenic, whereas enzymatic loss of function seems to be prevalent in myeloid disorders such as CML (98, 99).
In MM patients, EZH2 is often overexpressed and correlates with a more aggressive disease and shorter overall survival, independent of treatment modality. This overexpression causes transcriptional repression of cell cycle regulatory genes, leading to increased malignant proliferation (90). In accordance with this, the pharmacological inhibition of EZH2 in MM cell lines and primary cells confers increased drug sensitivity, inhibits cell growth and can lead to a reactivation of certain tumor-suppressive regulatory genes (100, 101). The role of EZH2 in hematological cancers is both stage- and lineage-dependent. Increased H3K27 methylation drives stemness and proliferation in early AML, B-cell lymphomas and MM, while loss of function can promote resistance in CML and treated AML. Although FDA-approved EZH2-targeted therapy is already in use for FL (102), we are yet to elucidate under which conditions and in which diseases its inhibition will have a successful effect.
Another lysine methyltransferase, Suppressor of Variegation 3–9 Homolog 1 (SUV39H1), is responsible for the maintenance of constitutive pericentric heterochromatin. As a result, repetitive DNA sequences are transcriptionally repressed and regulated (103). Dysregulation of SUV39H1 disrupts the balance of H3K9 methylation, promoting the development of hematological malignancies (104).
The function of SUV39H1 has been investigated mostly in AML, where it exerts both tumor suppressive and oncogenic roles (104, 105). Specifically, SUV39H1 expression was decreased in CD34^+^ AML cells compared to their healthy counterparts, resulting in an aberrant distribution of H3K9me3 (104). Deregulation of H3K9me3 in AML patients is well recognized and serves as a prognostic marker, when combined with clinical data (106). The repression of SUV39H1 and the resulting reduction of H3K9me3 leads to reduced repression of oncogenes, such as HOXB13, SIX1, and HOXA9, whose downstream targets drive self-renewal and proliferation. Restoring the expression of Suv39h1 halts disease progression and reduces the occurrence of leukemia stem cells in a murine model (104). The loss of SUV39H1 seems to sustain immature stem cell persistence by disrupting oncogene repression, enabling leukemic self-renewal and therapy resistance. Conversely, the activity of SUV39H1 in AML cells led to promoter histone methylation of tumor suppressor genes such as CDKN2B and E-cadherin (CDH1). The inhibition of the SUV39H1 enzyme restored their transcription. However, this study specifically investigated MLL-AF9 AML (MLL is fused with ALL1-fused gene from chromosome 9, AF9), which likely has a distinct epigenetic landscape that influences SUV39H1 behavior (107). In addition, the differences between these two studies are major and relate to varying experimental models (murine vs. primary samples harboring specific mutations), gene/enzyme repression strategies and investigative methodologies. This highlights the challenges in defining the precise function of a protein in a disease, yet doing so remains essential for developing effective therapies.
A similar pattern of epigenetic dysregulation is observed in MM, where nuclear receptor binding SET domain protein 2 (NSD2) or multiple myeloma SET domain protein (MMSET), is a histone methyltransferase overexpressed in cells with the t (4, 14) translocation (108). This results in a global increase of H3K36me2 and drastic gene expression alterations and is associated with a poor prognosis (108, 109). Due to the role of MMSET in the DNA damage response, this enzyme is also involved in treatment resistance in MM cells (110). Knockout or knockdown of MMSET in t (4, 14) MM cell lines leads to apoptosis, cell cycle arrest, and reduced tumor growth in vivo, highlighting the importance of this enzyme in MM cell survival (111). In MM cells with increased MMSET expression, a nota ble increase in EZH2 recruitment was observed, further contributing to myeloma development and survival (112). This reveals a broader pattern of cooperative epigenetic regulation involved in myeloma malignancy.
Histone demethylases are studied less in hematological cancers. However, overexpression of lysine-specific demethylase 1 (LSD1 or KDM1A) has been observed in AML cells (113). LSD1 is considered an important mediator of the differentiation block of MLL, characterized by rearrangements in the KMT2A gene and is correlated with a poor clinical outcome. This enzyme removes methyl groups from H3K4 and acts as part of a transcription repression complex (114). Through its activity, LSD1 represses the transcription of genes involved in myeloid differentiation, contributing to AML stemness.
In line with that, the inhibition of LSD1 leads to the differentiation of primary murine and human MLL cells, without affecting healthy hematopoietic progenitor cells (115). In addition, knockdown of LSD1 in AML cell lines led to a reduction in their clonogenic potential and proliferation. In vivo LSD1 knockdown also induced an upregulation of integrin alpha M (CD11b, ITGAM) and CD86 in AML, which are recognized myeloid differentiation markers (114). However, several studies showed the function of CD11b and CD86 in tumor progression (114, 116–118). Based on current knowledge, the role of CD11b is stage dependent – high baseline expression of CD11b indicates an aggressive leukemia phenotype, whereas therapy-increased CD11b is considered a good response, as it is associated with a differentiation block exit. Studies correlating the baseline expression of CD11b with a poor outcome in AML patients are mostly cohort studies and meta-analyses which do not discuss the molecular mechanisms behind this occurrence.
Likewise, LSD1 is overexpressed in other myeloid diseases, such as myeloproliferative neoplasms, CML, and MDS (119). The relevance of LSD1 in myeloid diseases suggests a shared dependency of epigenetic repression to differentiation and lineage fidelity.
In ALL, targeting LSD1 in a 3D culture setting increased the invasive capacity in cell lines, an effect opposite to that observed in vivo studies. In fact, LSD1 inhibition seems to have decreased the colonization potential of ALL cells in vivo, with impaired chemotaxis and migration (120). These findings emphasize the importance of contextual cues and the investigation of tumor cells in their native environment. Moreover, although useful, cell lines do not necessarily represent the original cancer ideally, and certain studies may therefore yield contrasting results.
Selective oncogenic pressure leads to an overexpression of histone methyltransferases that repress tumor suppressors and histone demethylases whose activity promotes the transcription of genes involved in self-renewal and proliferation. The activity of these enzymes is dictated by other proteins in the cells, which recruit them to specific sites. This explains the observed effects in cancer cells, but not in their healthy counterparts that have a different proteomic milieu. The transcriptional and proteomic profiles of these cells have not yet been investigated in such a systematic manner, but only correlated to genes involved in standardized cancer pathways – proliferation, apoptosis, self-renewal, etc. Therefore, gaps remain regarding a unified vision of how these processes mutually interact, emphasizing the need for integrative in vivo and clinical studies, combining them with current knowledge.
Histone acetylation in hematological malignancies
3.2
Histone acetylation is considered to activate transcription due to the loosening of chromatin structure, which makes DNA accessible to transcriptional regulators. Histone acetylation is mediated by two families of enzymes: histone acetyltransferases (HATs), which promote transcription, and histone deacetylases (HDACs), which repress transcription. The equilibrium between the activity of HATs and HDACs maintains normal hematopoietic differentiation, and its disruption contributes to hematopoietic malignancies (121). These enzymes are nowadays more often referred to as KATs and KDACs, to emphasize the fact they also (de)acetylize lysine residues on other proteins as well (122).
In hematological cancers, HATs are mutationally inactivated. For instance, a histone acetyltransferase gene, CREB-binding protein (CREBBP, or KAT3A encoding CBP/p300), was found to be mutated in 20% of relapsed ALL patients. These mutations were either present at diagnosis in certain subpopulations or acquired during relapse, indicating a correlation between HAT dysfunction and disease persistance. Observed CREBBP mutations affected the acetyltransferase domain, resulting in decreased H3K18 or H3K27 acetylation. This led to a dysregulated acetylation of promoters or enhancers of genes responsive to glucocorticoids, involved in the response to corticosteroid treatments, possibly rendering these cells resistant to the therapy (123).
Besides ALL, CREBBP mutations were found in other lymphoid malignancies, such as diffuse large B-cell lymphoma (DLBCL) and FL patients (124). This suggests a shared vulnerability of lymphoid-lineage cancers to dysregulated histone acetylation.
However, in myeloid malignancies, such as AML, the activity of CBR/p300 is linked to increased self-renewal, which can be reduced when an enzymatic inhibitor is applied (125). This dual role of CBR/p300 across different hematological malignancies may occur due to different lineage origins and differentiation stages. Either way, both CBR/p300 inhibition and restoration of activity can be beneficial, based on disease modality or mutational context, complicating future therapeutic strategies.
Overexpression of HDACs, rather than specific mutations, was detected in hematological malignancies. For example, HDAC1 was overexpressed in AML cells, which was linked to an oncogenic transcription regulation. HDAC1 negatively correlates with the Kruppel-like factor 4 (Klf4) tumor suppressor by competing for its promoter and inhibiting its expression. The reduction of Klf4 in turn reduces the expression cell cycle regulators, cyclin-dependent kinase inhibitors p21 (CDKN1A) and p27 (CDKN1B), and promotes proliferation. In addition, lower HDAC1 expression in AML patients was associated with longer overall and disease-free survival (126).
In addition to aberrant expression levels, HDACs are often recruited by pathogenic fusion proteins in acute leukemias. In AML and acute promyelocytic leukemia (APL) pathogenic fusion proteins are frequent and occur due to chromosomal translocations. The dysregulated engagement of HDACs leads to a suppression of tumor suppressor genes and, ultimately, to differentiation arrest and leukemia development (127, 128). While this general principle was established long ago, the specific tumor repressors targets and their contribution to pathogenesis are under-investigated.
HDAC-mediated transcriptional repression has been recognized in other hematological malignancies as well. Upregulation of HDACs is also observed in B-CLL cells, compared to healthy controls. These enzymes seem to play a complex role in CLL development, acting as both activators and repressors of transcription, depending on chromatin organization. Specifically, when HDAC1 is recruited to super-enhancers, it functions as a transcriptional activator of genes involved in CLL survival, such as B-cell lymphoma 2 (BCL2). Conversely, in regions without super-enhancer marks, HDAC1 represses transcription, particularly of tumor suppressor genes (129). This reveals a very high level of oncogene/tumor suppressor expression regulation, dependent on chromatin organization, since the proteins involved in guiding HDAC1 through other epigenetic marks also function in a protumorigenic manner. This study once again emphasizes the complexity of investigating the involvement of specific epigenetic regulators in one disease entity and underscores the necessity for a more complex, contextual, proteomic analysis.
In CML, HDAC overexpression is correlated with a drug-resistant population of cells and an increased expression of multipotent stemness genes such as SRY (sex-determining region Y)-box 2 (SOX2), homeobox transcription factor NANOG (NANOG), and octamer-binding transcription factor 4 (OCT4), which leads to disease persistence and treatment evasion (130).
In lymphomas, class I HDACs are broadly expressed and facilitate immune evasion and tumor suppressor gene silencing (131). Similarly to lymphomas, class I HDACs are dysregulated in MM, which is correlated with a poor prognosis (132).
Given the broadly established role of HDACs in lymphoid and myeloid malignancies, HDAC inhibitors such as vorinostat, belinostat, and romidepsin have already been approved for the treatment of hematological cancers as combinational therapies, but have failed to yield significant results (133).
Dysregulation of histone acetylation, whether through HAT mutations or HDAC overexpression plays an important role in hematological malignancies by regulating the expression of oncogenes and tumor suppressors. The effects of these enzymes are context dependent and vary by disease subtype.
Chromatin remodeling complexes in hematological malignancies
4
Chromatin remodeling refers to dynamic changes in chromatin structure, which interact with DNA methylation and histone modifications. It dictates the spatial organization of nucleosomes and, thus, the transcriptional availability of DNA. There are four major families of chromatin remodeling complexes, comprised of ATP-dependent enzymes: Switch/Sucrose Non-Fermenta ble (SWI/SNF), Imitation Switch (ISWI), Chromodomain-Helicase-DNA-Binding (CHD), and Inositol-Requiring 80 (INO80). Chromatin remodeling complexes are responsible for nucleosome repositioning, ejection, and restructuring which changes the accessibility of DNA for transcription (Figure 3) (134).
Chromatin remodeling complexes and their functions. Schematic representation of chromatin remodeling complexes SWI/SNF, ISWI, CHD, and INO80 that regulate nucleosome organization and chromatin accessibility. Chromatin remodeling proteins utilize ATP hydrolysis to organize the chromatin through nucleosome repositioning, ejection or sliding or nucleosome/histone exchange. The CHD family mediates nucleosome sliding and eviction, while ISWI complexes primarily regulate nucleosome spacing. SWI/SNF complexes are involved in nucleosome repositioning and ejection, opening chromatin. INO80 proteins facilitate histone exchange, including the replacement of canonical histones with histone variants. The activity of chromatin remodeling complexes regulates key DNA-dependent processes, including replication, transcription and DNA damage repair. Image created in Biorender.
SWI/SNF chromatin remodeling complexes in hematological malignancies
4.1
SWI/SNF complexes are comprised of numerous subunits and are involved in nucleosome repositioning and ejection, which opens chromatin and exposes promoter regions. Once recruited to DNA, specific histone modifications or transcription factors, the ATPase subunits utilize ATP hydrolysis energy to slide nucleosomes along DNA or eject them. This alters the chromatin structure, which either increases or restricts DNA accessibility. In this manner, SWI/SNF proteins modulate the ability of transcription factors and the RNA polymerase to interact with regulatory elements. SWI/SNF complexes regulate DNA replication, transcription, and genomic stability, guiding normal cellular development and differentiation. When these processes are dysregulated, they can contribute to the emergence of cancer. In hematology, SWI/SNF complexes function as tumor suppressors in lymphoid cancers via loss-of-function mutations, while acting as oncogenes in myeloid malignancies through gain-of-function mutations or overexpression (135).
In myeloid diseases, such as AML, mutations in SWI/SNF enzymes are rare, but their activity is correlated with cellular differentiation and patient survival. The SWI/SNF subunit SWI/SNF Related BAF Chromatin Remodeling Complex Subunit D1 (SMARCD1) is highly expressed in CD34^+^ hematopoietic cells, which decreases during regular myeloid differentiation. Increased SMARCD1 expression in AML patients suppresses myeloid differentiation via repression of transcription regulators of myeloid differentiation, such as CD11b, CD14 (monocyte/macrophage marker), S100A8/A9 (S100 calcium-binding proteins), and CCL3/4/7/8 (chemokine C-C-motif ligands). This repression is mediated by decreased histone methylation on H3K4 and increased histone methylation on H3K27. In this manner, SMARCD1 is involved in the differentiation block of AML, and its overexpression correlates with a stem cell signature and poor prognosis, promoting leukemogenesis and disease maintenance (136).
In contrast, in lymphoid malignancies, SWI/SNF complex proteins act as tumor suppressors via loss of function mutations. For instance, in DLBCL and FL, nonsense or frameshift alterations occur in the AT-rich interactive domain-containing protein 1A (ARID1A) subunit recurrently, truncating the protein (135, 137). In B cells, ARID1A chromatin remodeling activity assists the binding of transcription factors (such as nuclear factor kappa-light-chain-enhancer of activated B cells, NF-κB) to genes involved in B-cell germinal center differentiation and activation. Loss of ARID1A impairs B-cell maturation, influencing both patient immunity and lymphoma development (138).
Furthermore, in FL, nearly all patients carry mutations in chromatin remodeling genes, with ARID1A and SMARCA4 being the most common. It remains unclear why FL is particularly vulnerable to dysregulated chromatin remodeling. In fact, other epigenetic regulators, such as KMT2B, CREBBP and EZH2 are frequently mutated in this disease as well. It is speculated that FL’s dependence on epigenetic mutations stems from their germinal center (GC) B-cell origin, characterized by heavy proliferation and high mutational burden, with an increased tolerance for DNA damage. Thus, the mutations occurring in epigenetic regulators are tolerated as and can cumulatively give rise to malignant cells (139).
SMARCA4 mutations are very common in B-cell lymphomas as well. In germinal GC-derived Burkitt lymphoma, SMARCA4 mutations are found in 30% of patients. SMARCA4 is involved in regular GC B cell development. Monoallelic loss of SMARCA4 decreases the activity of transcription factors needed for GC exit, therefore disrupting normal B-cell development, which can ultimately lead to carcinogenesis. Recurrent mutations in SWI/SNF subunits such as ARID1B/2, SMARCA2 and SMARCA4 are observed in hematological cancers as well, indicating the important role these proteins have in oncogenesis. In addition, mutations in SWI/SNF genes were discovered in Burkitt lymphoma, Hodgkin lymphoma and other B, T and NK-cell lymphomas (135). These mutations are not equally distributed, but highlight a vulnerability of lymphoid cells to SWI/SNF dysregulation. However, it is still unclear whether these mutations initiate oncogenesis or emerge as secondary events, supporting neoplastic cells.
In MM, mutations in the BAF Chromatin Remodeling Complex Subunit 7A (BCL7A) SWI/SNF subunit gene were identified in non-coding regions in about 60% of patients. BCL7A is downregulated in MM cells compared to healthy plasma cells and is considered to have a tumor suppressive role, since loss of BCL7A led to increased MM growth and viability. BCL7A protein interacts with the Interferon Regulatory Factor 4 (IRF4) transcription factor to limit its function. Loss of BCL7A leads to the transcription of IRF4-targeted genes, which drives MM cell growth (140). Therefore, the loss (or lack) of BCL7A leads to IRF4 activation, promoting genes involved in MM proliferation. The specific target genes of IRF4 in MM are unknown. This study highlights the importance of mutations in non-coding regions, raising the question of their potential relevance in other epigenetic regulators, which might have been overlooked in whole-exome sequencing approaches thus far.
Mutations in SWI/SNF subunits are rare in leukemias, except for T-ALL, where BCL11B subunit mutations are speculated to be a driver of the disease and occurs fairly frequently – in about 10% of patients. BCL11B mutations are linked to all major subtypes of T-ALL, most commonly as monoallelic deletions or missense mutations. It is speculated that these mutations in T-ALL correlate with mammalian target of rapamycin (mTOR) signaling activation, generally involved in proliferation and survival, though exact mechanistic links are not described (141).
In summary, SWI/SNF dysregulation in hematological malignancies is involved in transcriptional changes, resulting in impaired differentiation and the maintenance of stemness in cancer cells. Despite the involvement of distinct subunits across different malignancies, these effects arise through diverse cellular mechanisms.
ISWI chromatin remodeling complexes in hematological malignancies
4.2
Subunits of the ISWI chromatin remodeling complex bind to nucleosomes and slide them along DNA, providing adequate nucleosome spacing, which limits chromatin accessibility and transcription (134, 142). ISWI complexes are comprised of either the SMARCA5 or SMARCA1 catalytic subunits, which translocate DNA through ATP hydrolysis, without disassembling the nucleosome (143, 144). This translocation occurs in a repetitive manner, through cycles of sliding and pausing, resulting in regularly spaced nucleosomes, which organizes the chromatin structure (145). Defects in ISWI function contribute to genomic instability and have been associated with cancer (146). In addition, the activity of ISWI complexes is required for proper hematopoietic differentiation and therefore essential for normal blood cell development (125).
In myeloid malignancies, altered ISWI activity is correlated with impaired differentiation and disease persistence. For example, SMARCA5, or Sucrose Non-Fermenting 2 Homolog (SNF2H), was overexpressed in AML CD34^+^ bone marrow cells, compared with their healthy counterparts, and was likely to contribute to differentiation dysregulation. When patients were in remission, the expression of SMARC5 was reduced, indicating a correlation between the enzyme with disease activity (147). The overexpression of SMARCA5 leads to an epigenetic repression in the myeloid differentiation regulator PU.1 in both AML cell lines and primary samples (148). Additionally, the deletion of SMARCA5 in AML cell lines led to cell cycle arrest, indicating its role in the maintenance of this cancer (149). Given that ISWI complexes are involved in the regulation of DNA repair, it is likely that the proliferation halt in these cells occurs due to the accumulation of DNA damage (144). Since ISWI complexes are responsible for genome integrity, their activity is crucial for cellular survival and proliferation.
In lymphoid malignancies, ISWI subunits exhibit oncogenic activity as well. For instance, Bromodomain Adjacent To Zinc Finger Domain 2A (BAZ2A), was overexpressed in CLL patients compared to respective healthy controls, due to the downregulation of tumor suppressor miRNAs (150). Even though the ISWI complex is essential for cellular integrity, the overexpression of SMARCA5 in AML patients and BAZ2A in CLL patients indicates their oncogenic role, as it might influence the well/calibrated proteomic balance in the cell and lead to dysregulation in genome organization.
The overexpression of ISWI subunits is found in both AML and CLL, despite their myeloid and lymphoid origins, suggesting a shared oncogenic mechanism. Although these studies identified ISWI protein involvement in several hematological cancers, mechanistic input or clinical relevance are not precisely described. There aren’t many studies characterizing the function of the ISWI complex in other hematological malignancies. However, its role in hematopoiesis and its oncogenic function suggests relevance in these cancers and warrants further investigation.
CHD chromatin remodeling complexes in hematological malignancies
4.3
The CHD family of proteins organizes histone octamers into stable nucleosomes upon replication. They are involved in nucleosome ejection or sliding, as well as histone deacetylation. The interaction of CHD proteins with modified histone tails, DNA, or transcription factors leads to the activation of their ATPase activity, which in turn slides or repositions nucleosomes. CHD proteins use ATP hydrolysis to twist the DNA around the histones, which changes DNA accessibility and transcription factor binding, regulating transcription. It is uncertain whether CHD proteins directly interact with other epigenetic proteins, such as DNMTs or TETs, but they do depend greatly on their activity, as they are recruited through the recognition of specific histone modifications (151).
Their role in hematological cancers has not been investigated thoroughly. Recent studies show that certain members of this family, such as CHD4, CHD7, and CHD9 are overexpressed in AML patients, suggesting their tumor promoting roles in myeloid lineage cells (152). CHD4 regulates the expression of the MYC oncogene in AML, promoting cell cycle progression Additionally, shRNA knockdown of CHD4 in AML cell lines opens the chromatin, increasing its susceptibility to double-stranded breaks when genotoxic treatments are applied (153). The correlation between the expression of CHD proteins and AML patient clinical parameters suggests their relevance in this disease, but the data lacks conviction (152).
In contrast, CHD2 gene was mutated in CLL patients, suggesting its tumor suppressive role. CHD2 mutations result in reduced association with chromatin, leading to transcriptional deregulation of genes involved in DNA damage repair and lymphocyte differentiation. These mutations often coincide with mutations in immunoglobulin heavy chain variable region genes, which are correlated to a better prognosis and usually indicate indolent disease. This raises the question of the clinical relevance of CHD mutations in CLL, which remains to be elucidated (154).
In leukemias, CHD5 seems to have a tumor suppressive role. CHD5 is silenced via promoter hypermethylation in CML, suggesting a cooperative link between different epigenetic mechanisms to promote leukemogenesis. In CLL cells, CHD5 restoration led to cell cycle arrest and promoted apoptosis due to the upregulation of the p21 cell cycle regulator. In vivo, CHD5 overexpression reduced cancer cell growth (155).
Limited data suggest that certain CHD proteins may function as oncogenes in plasma cell malignancies. One in vitro study was conducted regarding the involvement of CHD1L in MM. It was observed that CHD1L has an anti-apoptotic function in MM cell lines and that its overexpression led to increased cell adhesion/mediated drug resistance. This suggests that CHD1L might contribute to drug resistance in MM. However, this study was only conducted on cell lines, and more investigation is warranted to confirm these findings (156).
INO80 chromatin remodeling complexes in hematological malignancies
4.4
INO80 chromatin remodeling complex is involved in histone sliding and ejection, as well as histone exchange. By repositioning or evicting nucleosomes, INO80 proteins modulate chromatin organization, which influences replication, transcription and DNA repair. INO80 exchanges the histone variant H2A.Z with canonical H2A in nucleosomes, which changes promoter and enhancer accessibility (157). Specifically, H2A.Z-containing nucleosomes are less stable, resulting in a more plastic chromatin structure. This is associated with transcriptional activation and a high responsiveness to molecular signaling (158). Several studies have observed increased H2A.Z chromatin presence in various cancers, contributing to tumor growth, drug response, and metastasis (159–161).
The evidence about INO80 involvement in hematological malignancies is scarce and their role in these cancers remains speculative. Transcriptomic data identified mutations of INO80 in DLBCL, along with other genes whose protein products are linked to chromatin maintenance and regulation, such as EZH2, CREBBP, TET2, DNMT3A and others (162). It seems that epigenetic dysregulation is a hallmark of DLBCL. No mechanistic or clinically relevant data were acquired regarding INO80 proteins in this cancer. Given the vulnerability of hematological malignancies to epigenetic dysregulation, INO80 is likely implicated in their biology, although this remains uninvestigated.
Overall, chromatin remodeling complexes regulate gene accessibility and expression. Dysregulation of SWI/SNF, ISWI, CHD, and INO80 complexes leads to impaired differentiation, increased stemness, and the activation of oncogenic transcriptional programs in hematological malignancies. While SWI/SNF complexes often harbor mutations in lymphoid malignancies and are overactivated in myeloid malignancies, other complexes exhibit context-dependent roles. Given their ubiquitous expression, therapeutic targeting seems unlikely. Nonetheless, a systematic investigation into their interaction with other epigenetic regulators is essential in understanding cancer cell plasticity and adaptability. In this manner, we would identify additional vulnerabilities that could be therapeutically exploited indirectly.
Interplay between epigenetic mechanisms in hematological cancers
5
Epigenetic mechanisms act as a complex network of proteins that regulate gene expression. Coordinated crosstalk between DNA methylation, histone modifications and chromatin organizing complexes defines all cellular functions. Their dysregulation can promote tumor development and growth (Figure 4). Although many studies have investigated numerous epigenetic regulators in hematological cancers, it is still unclear how exactly their interaction functions to orchestrate neoplastic transformation.
Integrated model of epigenetic dysregulation in hematological cancers. Changes in DNA methylation enzymes, structure of chromatin, histone modification enzymes, and chromatin remodeling complexes act synergistically to contribute to the development of hematological cancers. These alterations often reinforce one another through positive feedback loops, resulting in widespread epigenetic dysregulation. For instance, dysregulation of DNA methylation proteins leads to altered DNA methylation patterns. This influences the recruitment of histone modifiers, changes chromatin architecture and chromatin organization complex activity. The combined effects of these alterations contribute to key oncogenic processes, such as blocked differentiation, tumor suppressor repression, oncogene activation, and drug resistance, which together promote oncogenesis and persistence of hematological cancers. Red crosses indicate dysregulation. Image created in Biorender.
Distinct DNA methylation patterns recruit DNMTs in a specific, context-dependent manner. In hematological cancers, DNMT3A mutations are common, which cause genome-wide hypomethylation, removing these marks and disturbing regular DNA methylation patterns. This modulates the function of histone methyltransferases, whose activity is also dependent on DNA methylation (163, 164). For instance, in AML, mutations in DNA methylation proteins such as DNMT3A, IDH1/2, or TET2 enzymes often co-occur and cooperate to drive leukemogenesis (41). IDH2 mutations are also linked to histone hypomethylation in AML, which influences the activity of histone methyltransferases (163). In ALL, the histone methyltransferase KDM2B is upregulated, acting cooperatively with HATs to induce oncogene expression (165). Increased MMSET expression in MM cells is associated with enhanced EZH2 recruitment, supporting myeloma progression and survival (112). Data about interactions of epigenetic modulators converging on specific genes is scarce, limiting our understanding of how these layers of regulation interact to control oncogenic and tumor-suppressive programs. Taken together, the deregulation of DNA methylation, histone modification, and chromatin remodeling complexes are observed in hematological malignancies. We are yet to determine how exactly all of them interact with each other to obtain a clear picture of disease pathogenesis and maintenance. The changes in DNA methylation influence histone modification and vice versa. This, in turn, changes the chromatin structure and DNA accessibility which further dictates the activity of these enzymes and chromatin remodeling complexes and their mutual interactions.
To systematically present epigenetic changes in hematological cancers, their summary was provided in Table 1.
The overarching epigenetic patterns in hematological malignancies involve suppression of cell differentiation, leading to more immature, less differentiated phenotypes. Furthermore, epigenetic regulation and chromatin architecture repress tumor suppressors and promote oncogene expression. This promotes cancer cell survival, resulting in more drug-resistant clones with the ability to relapse. Given the dynamic nature of epigenetic marks, this occurs due to cumulative selection of cells with adaptive behavior during cancer development and subsequent therapeutic pressure.
Clinical translation and therapeutic implications of epigenetic alterations
6
Most epigenetic therapeutics approved thus far are used in hematological malignancies, due to the relevance of epigenetic dysregulation in these cancers. Currently approved FDA therapies involve DNMT, HDAC, IDH, and EZH2 inhibitors (167).
DNMT inhibitors azacitidine and decitabine, which target DNMT1 primarily, were among the first epigenetic drugs approved and are currently used in the treatment of AML, MDS and CML. These drugs induce hypomethylation in cancer cells, which can lead to the expression of silenced tumor suppressor genes. However, hypomethylation caused by these inhibitors does not correlate with clinical outcome, and is often transient, resulting in attenuated therapeutic efficacy (168, 169). Rapid restoration of methylation patterns, upregulation of DNMT1 and DNMT3B and metabolic drug manipulation can all contribute to resistance to DNMT inhibitors (170). Even though DNMT enzymes are upregulated in many hematological cancers and used as monotherapy in some cases, their sole inhibition does not eliminate cancer cells long-term. The question arises whether the problem is their activity per se, or rather a dysregulated distribution of DNA methylation that needs to be restored.
HDAC inhibitors vorinostat, romidepsin, belinostat, and panabinostat increase histone acetylation which opens chromatin and reactivates silenced tumor suppressor genes, which can lead to cell cycle arrest and apoptosis (171). Vorinostat is approved for relapsed/refractory CTCL; romidepsin for CTCL and peripheral T−cell lymphoma (PTCL); belinostat for relapsed/refractory PTCL; and panobinostat, in combination with bortezomib and dexamethasone, for relapsed/refractory MM (167). HDAC inhibitors used as monotherapy have failed to yield significant results, since patients rarely achieve complete remission or long-term disease-free survival (171, 172). The insufficient efficacy of these therapeutics could be attributed to various factors. HDACs deacetylate other proteins as well, involved in apoptosis, DNA repair, and survival. The inhibition of HDACs increases acetylation on other proteins, which can result in their (increased) activation. This can provide an additional advantage to cancer cells by promoting broader resistance phenotypes. In addition, cancer cells treated with HDAC inhibitors can upregulate anti-apoptotic proteins, increase DNA damage response, or employ antioxidant defense pathways (173).
FDA-approved IDH inhibitors are enasidenib, ivosidenib and olutasidenib, used for the treatment of AML patients with recognized IDH mutations. These drugs specifically target mutated IDH1 or IDH2 proteins. They lovers 2-HG levels in the cells, restoring TET function and releasing the differentiation block. These therapies represent a good example of precision medicine, where only mutated isoforms are targeted, minimizing the therapeutic influence on healthy cells. Since their efficacy as monotherapies has proven insufficient, they are planned to be integrated into combination therapies (174). Given that these therapeutics have been recently approved, not much is known about the mechanisms behind drug response. However, in AML treated with IDH inhibitors, leukemia cells are sometimes found to switch between IDH1 or IDH2 mutations at relapse, evading targeted therapy (175).
EZH2 inhibitor tazemetostat is used in the treatment of FL. The inhibition of EZH2 leads to reduced H3K27 methylation and derepression of differentiation-related and tumor suppressor genes. Tazemetostat inhibits both mutant and wild−type EZH2, leading to cell cycle arrest, apoptosis, and inhibited tumor growth in lymphoma cells. Thus far, tazemetostat seems to be well-tolerated and effective, with a potential to be used in combination therapies for improved clinical outcomes (102).
Other epigenetic drugs are currently under investigation as well. For example, inhibitors of the LSD1 histone demethylase are under investigation for the treatment of AML. LSD1 inhibitors promote differentiation in AML cells by increasing H3K4 methylation. They often only induce partial myeloid differentiation, and their activity can be compensated by other histone demethylases, which is why they exhibit limited treatment success as monotherapy. Therefore, combination therapies are under investigation, including strategies targeting signaling pathways involved in the maintenance of stemness (176). Development of novel epigenetic inhibitors is ongoing, generating next-generation drugs, with enhanced efficacy or targeting new epigenetic proteins altogether. Taken together, there is considerable potential in targeting epigenetic regulators, which clearly contribute to hematological cancers. To do so efficiently, a deeper understanding of epigenetic modalities is required—in both disease- and stage- specific context and their reciprocal interactions. It is likely that epigenetic drugs will not be effective as monotherapy but will need to be used in combination with other therapies to achieve meaningful clinical outcomes.
In addition, epigenetic biomarkers have emerged as powerful tools in the diagnostics, prognosis, and treatment of hematological malignancies. DNA methylation patterns define epitypes and methylation classifiers, which provide improved prognostic prediction. For instance, specific CpG island methylation signatures in AML, lymphoma, and MM have been shown to correlate with treatment response and survival outcomes (64, 66, 67, 74, 75). Integrating epitypes into clinical workflows is a good example of combining epigenetic insights into clinical practice and therapeutic strategies.
The future therapeutic use of epigenetic knowledge should integrate the development of drugs that precisely target relevant epigenetic regulators, together with the use of predictive models for patient stratification and precision medicine. The combination of epigenetic drugs with other therapeutical modalities may enhance efficacy and overcome resistance. Advances in patient-specific epigenetic profiling will enable personalized treatment strategies, improving outcomes while minimizing toxicity.
Conclusion
7
DNA methylation, histone modifications and chromatin remodeling are involved in the development and maintenance of hematological malignancies. Epigenetic gene expression regulation facilitates swift responses to alternating cellular signals, enabling a quick adaptation of the cell to changing environmental cues. This plasticity is exploited extensively by cancer cells contributing to treatment evasion and clonal evolution and ultimately, drug resistance and relapse. Therefore, future frameworks should integrate epigenetic regulation into cancer research, investigated with multi-omic approaches, AI predictive models, and chromatin conformation analysis to uncover the drivers of hematological malignancies and therapeutic resistance, offering significant potential for new discoveries.
Given the role epigenetic regulators clearly play in hematological malignancies, numerous epigenetic inhibitors are used for their treatment. Clinically approved epigenetic inhibitors are used in the treatment of hematological cancers, often in combination with other therapies, but their efficacy remains limited. In addition, it can be particularly challenging to target epigenetic regulators, due to their ubiquitous function. A more selective approach, such as targeting specifically mutated enzymes, could provide more favorable outcomes in patients in the future.
Even though epigenetic inhibitors are widely used in the treatment of hematological cancers, they have largely failed to achieve desired clinical outcomes. Recent studies of hematological cancers focus on immunotherapies and DNA mutational events, while overlooking the epigenetic context. A comprehensive understanding of these dynamic regulatory changes, their interactions, and integration into novel treatment approaches, is necessary to further improve treatment. Most studies investigate one particular epigenetic mark, due to obvious experimental limitations. However, it would be of significant value to establish libraries of specific histone code patterns, chromatin accessibility and DNA spatial organization, and to map their relevance in cancer progression. Future approaches should focus on providing a more integrative and systematic investigation in this field. For example, AI-based predictive frameworks could correlate chromatin conformation states with transcriptional outputs. This could predict how combinations of histone modifications shape enhancer–promoter interactions and identify epigenetic determinants of immunotherapy resistance. In addition, predictive AI models might reveal connections between epigenetic patterns and chromatin conformation states with proteomic and transcriptomic outputs. This can uncover epigenetic determinants and provide insights into specific cancer behaviors. In addition, data acquired from multi-omics approaches, such as ATAC-seq, ChIP-seq and single cell RNA-seq could help identify novel vulnerabilities, (epigenetic) cell heterogeneity and microenvironmental dependencies.
The extensive amount of data already collected has enormous potential to yield more definitive conclusions when summarized and put into a biological context by computational methods. Finally, the integration of discovered epigenetic markers could be utilized in precision medicine, by developing patient-specific profiles which could predict and improve treatment responses.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lachmann M Libby E . Epigenetic inheritance systems contribute to the evolution of a germline. Philos Trans R Soc Lond B Biol Sci. (2016) 371:20150445. doi: 10.1098/rstb.2015.0445, PMID: 27431523 PMC 4958939 · doi ↗ · pubmed ↗
- 2Jaenisch R Bird A . Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Gen. (2003) 33:245–54. doi: 10.1038/ng 1089, PMID: 12610534 · doi ↗ · pubmed ↗
- 3Holoch D Moazed D . RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. (2015) 16:71–84. doi: 10.1038/nrg 3863, PMID: 25554358 PMC 4376354 · doi ↗ · pubmed ↗
- 4Bachireddy P Burkhardt UE Rajasagi M Wu CJ . Haematological Malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. (2015) 15:201–15. doi: 10.1038/nrc 3907, PMID: 25786696 PMC 4511812 · doi ↗ · pubmed ↗
- 5Orkin SH Zon LI . Hematopoiesis: an evolving paradigm for stem cell biology. Cell. (2008) 132:631–44. doi: 10.1016/j.cell.2008.01.025, PMID: 18295580 PMC 2628169 · doi ↗ · pubmed ↗
- 6Zhao A Zhou H Yang J Li M Niu T . Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic Malignancies. Signal Transduct Target Ther. (2023) 8:71. doi: 10.1038/s 41392-023-01342-6, PMID: 36797244 PMC 9935927 · doi ↗ · pubmed ↗
- 7Jin B Robertson KD . DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. (2013) 754:3–29. 22956494 10.1007/978-1-4419-9967-2_1PMC 3707278 · doi ↗ · pubmed ↗
- 8Gao L Emperle M Guo Y Grimm SA Ren W Adam S . Comprehensive structure-function characterization of DNMT 3B and DNMT 3A reveals distinctive de novo DNA methylation mechanisms. Nat Commun. (2020) 11:3355. doi: 10.1038/s 41467-020-17109-4, PMID: 32620778 PMC 7335073 · doi ↗ · pubmed ↗