From Childhood Icterus to Adolescent Gallstones: Clinically Diagnosed Crigler‐Najjar Syndrome Type II
Danish Kumar Goswami, Barkha Goswami, Samiullah Shaikh, Abdul Ghani Rahimoon, Rakesh Soni, Kamil Ahmad Kamil

TL;DR
A young man with lifelong jaundice and gallstones was diagnosed with a rare liver metabolism disorder using a drug response test.
Contribution
Demonstrates phenobarbital responsiveness as a diagnostic tool for CNS-II in settings without genetic testing.
Findings
Phenobarbital therapy significantly reduced bilirubin levels, confirming CNS-II diagnosis.
Chronic hyperbilirubinemia led to gallstone formation in a CNS-II patient.
Phenobarbital responsiveness can serve as a diagnostic marker in resource-limited settings.
Abstract
Crigler–Najjar syndrome type II (CNS‐II) is an uncommon cause of persistent unconjugated hyperbilirubinemia resulting from partial deficiency of hepatic UDP‐glucuronosyltransferase activity. We report the case of a 19‐year‐old male who presented with intermittent jaundice since childhood and recent worsening of scleral icterus. Laboratory evaluation revealed isolated unconjugated hyperbilirubinemia with normal liver enzymes. Genetic testing was unavailable; however, serum bilirubin levels declined significantly following phenobarbital therapy, confirming the diagnosis of CNS‐II. Abdominal ultrasonography demonstrated gallstones, indicating chronic bilirubin supersaturation secondary to longstanding hyperbilirubinemia. The patient was managed conservatively with phenobarbital and counseling on avoiding precipitating factors such as fasting and hepatotoxic drugs. This case underscores the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
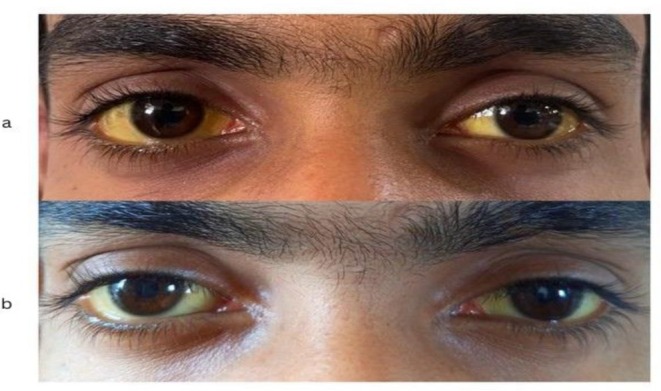 FIGURE 1
FIGURE 1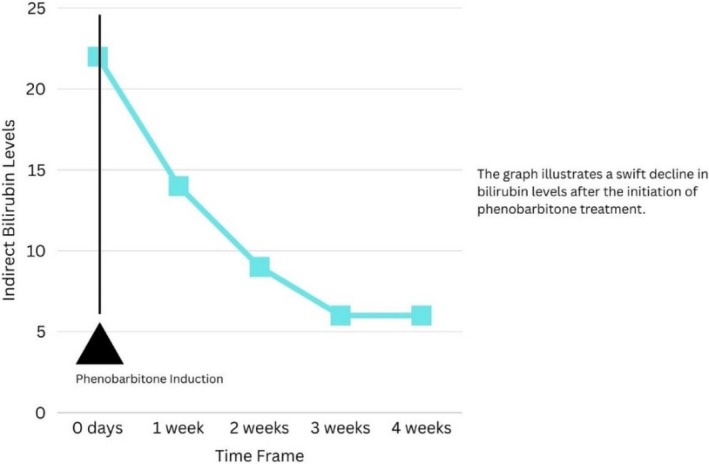 FIGURE 2
FIGURE 2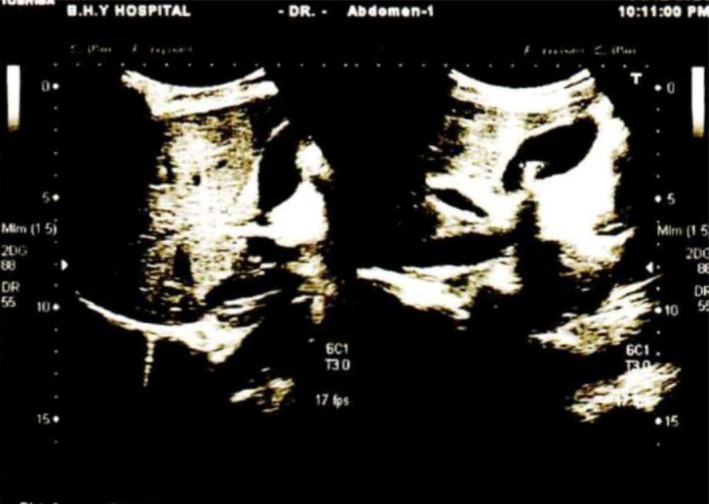 FIGURE 3
FIGURE 3| Parameter | Result at presentation (baseline) | Follow‐up after phenobarbital (4 weeks) |
|---|---|---|
| Hematology | ||
| Hemoglobin | 14.2 | 14.3 |
| Total leukocyte count | 7.4 | 7.1 |
| Platelet count | 265 | 252 |
| Liver function tests (LFTs) | ||
| Total bilirubin (mg/dL) | 23.51 | 6.2 |
| Indirect (unconjugated) bilirubin (mg/dL) | 22.86 | 5.59 (≈70% reduction from baseline) |
| Direct (conjugated) bilirubin (mg/dL) | 0.65 | 0.61 |
| AST (U/L) | 26 | 25 |
| ALT (U/L) | 29 | 33 |
| ALP (U/L) | 90 | 78 |
| INR/prothrombin time | 0.9 | 1.0 |
| Renal function and electrolytes | ||
| Serum creatinine | 0.6 | — |
| Blood urea nitrogen | 18 | — |
| Serum electrolytes (Na+, K+, Cl−) | Within normal limits | — |
| Viral serology | ||
| HBsAg | Negative | — |
| Anti‐HCV | Negative | — |
| HIV | Negative | — |
| Hemolysis workup | ||
| Reticulocyte count | 1.0 | — |
| Direct Coombs test | Negative | — |
| LDH | 90 | — |
| Serum haptoglobin | 120 | — |
| G6PD level | Within normal limits | — |
| Hemoglobin electrophoresis | Normal pattern; no hemoglobinopathy | — |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeonatal Health and Biochemistry · Pediatric Hepatobiliary Diseases and Treatments · Methemoglobinemia and Tumor Lysis Syndrome
Introduction
1
Crigler–Najjar syndrome (CNS) is a rare autosomal recessive disorder of bilirubin metabolism caused by mutations in the uridine diphosphate‐glucuronosyltransferase (UGT1A1) gene, leading to reduced or absent activity of the hepatic enzyme UGT1A1, thus resulting in unconjugated hyperbilirubinemia of varying severity. CNS is classified into two types: type I, which is severe and presents in the neonatal period with complete enzyme deficiency, and type II, which is milder and presents in late childhood or adolescence, characterized by partial enzyme activity and phenobarbital responsiveness [1, 2].
Patients with CNS‐II may develop complications such as cholelithiasis due to chronic bilirubin supersaturation and calcium–bilirubinate stone formation [3]. The global prevalence is estimated at approximately 0.6–1 per million live births [1], though the incidence is higher in regions with prevalent consanguineous marriages, such as South Asia [4]. Phenobarbital, a cytochrome P450 inducer, enhances the remaining activity of the UGT1A1 enzyme and has been shown to improve the clinical manifestations of the disease [5]. In many low‐resource settings, UGT1A1 gene testing is not readily available, and diagnosis relies on biochemical profiling and the documented reduction in unconjugated bilirubin after phenobarbital therapy [6].
We report a rare case of a young adult male with CNS‐II, complicated by gallstone formation, diagnosed clinically based on persistent unconjugated hyperbilirubinemia and a marked phenobarbital response. This case underscores the diagnostic value of a supervised phenobarbital trial and highlights the need to recognize CNS‐II in adolescents with long‐standing jaundice. This case is described in accordance with CARE guidelines [7].
Case Presentation
2
Presenting Complaints and Disease History
2.1
A 19‐year‐old male student with no history of tobacco use, alcohol consumption, or recreational drug use presented to the emergency department with right upper quadrant abdominal pain and persistent yellow discoloration of the eyes. On further inquiry, he reported a history of jaundice and cholelithiasis since the age of five. He noted that the icterus tended to worsen during periods of stress and intercurrent illnesses.
Despite multiple hospital admissions over the years and various conventional and alternative treatments, no definitive diagnosis had been established. His birth history was unremarkable, with no history of neonatal jaundice or need for phototherapy or exchange transfusions, and all developmental milestones were achieved on time. There was a positive family history of consanguinity in his parents, and one paternal cousin had also reportedly suffered from jaundice since birth. Based on these findings, the patient was admitted to the medical ward for detailed evaluation and further investigations.
On physical examination, the patient had marked scleral icterus without peripheral stigmata of chronic liver disease (Figure 1a). The abdomen was soft and nontender, with no organomegaly, and the remainder of the systemic examination was unremarkable.
Clinical manifestation of a patient showing scleral icterus. (a) Marked scleral icterus in a 19‐year‐old male with long‐standing unconjugated hyperbilirubinemia before phenobarbital therapy. (b) Same patient four weeks after phenobarbital therapy, showing visibly reduced scleral icterus.
Lab Investigation and Imaging
2.2
Initial laboratory investigations revealed a total serum bilirubin of 23.51 mg/dL, with an indirect (unconjugated) fraction of 22.86 mg/dL and a direct (conjugated) fraction of 0.65 mg/dL (Table 1). Other liver function tests were within normal limits, and there was no evidence of coagulopathy. Complete blood count, renal function tests, and serum electrolytes were all within normal ranges. Viral serology for hepatitis B and C, as well as HIV, was negative. A comprehensive hemolysis workup, including direct Coombs test, lactate dehydrogenase (LDH), serum haptoglobin, and glucose‐6‐phosphate dehydrogenase (G6PD) levels, was unremarkable (Table 1). Hemoglobin electrophoresis showed normal hemoglobin patterns with normocytic, normochromic red blood cells. Abdominal ultrasonography demonstrated cholelithiasis within the gallbladder lumen without biliary ductal dilatation or obstruction (Figure 3).
In view of isolated unconjugated hyperbilirubinemia with normal liver enzymes and no evidence of hemolysis, the main differentials considered were Gilbert syndrome and Crigler–Najjar syndrome type II (CNS‐II). Gilbert syndrome was considered less likely for two reasons. First, the patient's markedly elevated indirect bilirubin (22.86 mg/dL), which exceeds the typical levels (< 4.93 mg/dL) seen in Gilbert syndrome [8]. Second, the patient's long‐standing history of jaundice from early childhood, with multiple hospital visits, was more suggestive of CNS‐II than of Gilbert syndrome, which manifests as mild, intermittent episodes of scleral icterus or is discovered incidentally on blood tests, and usually does not lead to significant morbidity or repeated hospitalizations [9]. In our resource‐limited healthcare setting, genetic testing for UGT1A1 mutations was not available and the patient's family declined private genetic testing due to considerable financial burden. The diagnosis of CNS‐II was therefore made on clinical grounds, biochemical profile, and the documented response to phenobarbital (Figure 2).
Serial trend in serum bilirubin demonstrating a marked decline in unconjugated (indirect) bilirubin following initiation of phenobarbital therapy, with approximately 50% reduction at 1 week and nearly 70% reduction at 4 weeks compared with baseline.
Abdominal ultrasonography showing the gallbladder in longitudinal view with multiple echogenic intraluminal calculi producing posterior acoustic shadowing. The adjacent liver appears normal in echotexture, with no sonographic evidence of biliary ductal dilatation.
Therapeutic Intervention
2.3
Given the high unconjugated bilirubin levels and clinical suspicion of CNS‐II, a therapeutic trial of oral phenobarbital was initiated. Phenobarbital was started empirically at a total daily dose of 180 mg, administered in two divided doses, after detailed counseling regarding potential risks and benefits. Liver function tests and bilirubin levels were monitored weekly following initiation of therapy.
Within 1 week, the patient's unconjugated bilirubin decreased by approximately 50%, indicating a significant response to phenobarbital (Figure 2). This phenobarbital responsiveness, in combination with the biochemical and clinical profile, supported the diagnosis of CNS‐II.
Follow‐Up and Outcomes
2.4
The patient was followed up after 4 weeks of therapy. His indirect bilirubin level decreased from 22.86 to 5.59 mg/dL and remained stable at this level thereafter, representing an overall reduction of nearly 70% from baseline (Figure 2). Clinically, he reported improvement in jaundice and overall well‐being (Figure 1b).
Based on the clinical course, laboratory trend, and robust response to phenobarbital, the diagnosis of CNS‐II was confirmed. After appropriate counseling regarding the benign but lifelong nature of the condition, avoidance of precipitating factors (such as fasting and hepatotoxic drugs), and the risk of pigment gallstone complications, the patient was referred to the surgical department for further evaluation and management of his cholelithiasis. The patient subsequently underwent an elective laparoscopic cholecystectomy, which revealed multiple pigment stones without biliary ductal dilatation. His postoperative course was uneventful. At 6‐month follow‐up, he remained asymptomatic from a biliary standpoint and continued low‐dose phenobarbital (60 mg once daily), with stable unconjugated bilirubin around 5–6 mg/dL and normal liver enzymes.
Discussion
3
Although the biochemical profile and phenobarbital responsiveness of our patient are typical for CNS‐II, this case is notable for three reasons: presentation in late adolescence after years of undiagnosed jaundice, the occurrence of symptomatic cholelithiasis at a young age, and confirmation of CNS‐II using a structured phenobarbital trial in the absence of genetic testing. These features underscore practical diagnostic and management challenges in resource‐limited settings.
Our patient's diagnostic workup demonstrated isolated, marked unconjugated hyperbilirubinemia with normal liver enzymes, normal synthetic function, and a negative hemolysis workup, fulfilling the classic biochemical pattern of CNS‐II (Table 1) [10]. In many reports, CNS‐II has been diagnosed on a clinical and biochemical basis, with phenobarbital responsiveness used as a functional test of residual UGT1A1 activity when genetic testing is unavailable [4, 11, 12]. Moreover, phenobarbital induction therapy can be used to clinically differentiate CNS‐I, CNS‐II, and Gilbert syndrome. In CNS‐II, phenobarbital induces residual UGT1A1 activity and typically produces a ≥ 25%–50% reduction in serum unconjugated bilirubin, whereas patients with CNS‐I show little or no change [13]. By contrast, patients with Gilbert syndrome have much lower baseline bilirubin levels, and phenobarbital commonly normalizes or near‐normalizes their bilirubin levels [14]. In practice, a supervised trial of phenobarbital for 1–2 weeks at standard doses, with careful monitoring of bilirubin trends and adverse effects, provides both diagnostic confirmation and therapeutic benefit in CNS‐II. In our patient, unconjugated bilirubin fell by nearly 70%, with clear improvement in scleral icterus, which provides strong functional evidence of CNS‐II, closely mirroring the response profile reported in other CNS‐II cases diagnosed without immediate access to genetic testing [4, 15]. The presence of gallstones in this patient is biologically and clinically relevant. Chronic unconjugated hyperbilirubinemia promotes calcium–bilirubinate precipitation and black pigment gallstone formation, making gallstone disease an under‐recognized complication of CNS‐II and other unconjugated hyperbilirubinemias [16]. In our case, ultrasound‐confirmed cholelithiasis explained his biliary symptoms, and elective cholecystectomy (with ongoing low‐dose phenobarbital) provided effective combined management.
This case has broader implications for clinicians practicing in diverse healthcare environments. In resource‐limited health systems, where UGT1A1 sequencing is not routinely available, a stepwise, clinically driven approach, documenting isolated unconjugated hyperbilirubinemia, excluding hemolysis and liver disease, considering family history and consanguinity, and performing a supervised phenobarbital trial, can prevent diagnostic delay and unnecessary, repeated investigations [4]. At the same time, in settings where genetic testing is readily available, knowing that typical lab findings, along with a clear drop in bilirubin after phenobarbital, strongly point to CNS‐II can help clinicians order genetic tests more selectively, reserving it for equivocal cases or family counseling; thus, reducing costs [6].
The main limitation of this report is the absence of a confirmatory UGT1A1 genetic analysis. However, this limitation reflects a common reality in many low‐ and middle‐income regions, where the cost and availability of molecular testing remain significant barriers. Despite this constraint, our case adds value by illustrating a practical, reproducible diagnostic pathway for CNS‐II, highlighting the risk of early pigment gallstone disease, and emphasizing that a structured phenobarbital trial remains a powerful diagnostic and therapeutic tool where genomic confirmation is not readily available.
Conclusion
4
This case illustrates a rare presentation of CNS‐II in a young adult with lifelong unconjugated jaundice complicated by pigment gallstones. It highlights the need to consider CNS‐II in persistent unconjugated hyperbilirubinemia and supports the use of a supervised phenobarbital trial as a practical diagnostic and therapeutic tool, particularly where molecular testing is limited and to promote judicious use of genotyping when it is available. Early recognition, appropriate counseling, and long‐term follow‐up can prevent unnecessary investigations, enable timely management of biliary complications, and improve patients' quality of life.
Author Contributions
Danish Kumar Goswami: conceptualization, investigation, writing – original draft. Barkha Goswami: methodology, writing – original draft. Samiullah Shaikh: conceptualization, writing – review and editing. Abdul Ghani Rahimoon: validation. Rakesh Soni: data curation. Kamil Ahmad Kamil: investigation.
Funding
The authors have nothing to report.
Ethics Statement
The authors have nothing to report.
Consent
We explained everything to the patient in his native language, Sindhi, and obtained written informed consent for the publication of this case as per Wiley guidelines.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1J. Bhandari , P. K. Thada , M. Shah , and D. Yadav , Crigler‐Najjar Syndrome. Stat Pearls [Internet] (Stat Pearls Publishing, 2025), http://www.ncbi.nlm.nih.gov/books/NBK 562171/.32965842 · pubmed ↗
- 2P. L. Jansen , “Diagnosis and Management of Crigler‐Najjar Syndrome,” European Journal of Pediatrics 158, no. Suppl 2 (1999): S 89–S 94, 10.1007/pl 00014330.10603107 · doi ↗ · pubmed ↗
- 3S. R. Fernandes , C. M. Moura , B. Rodrigues , L. A. Correia , H. Cortez‐Pinto , and J. Velosa , “Acute Cholangitis in an Old Patient With Crigler‐Najjar Syndrome Type II ‐ a Case Report,” BMC Gastroenterology 16 (2016): 33, 10.1186/s 12876-016-0449-9.26968162 PMC 4788912 · doi ↗ · pubmed ↗
- 4A. Liaqat , A. Shahid , H. Attiq , A. Ameer , and M. Imran , “Crigler‐Najjar Syndrome Type II Diagnosed in a Patient With Jaundice Since Birth,” Journal of the College of Physicians and Surgeons‐Pakistan JCPSP 28 (2018): 806–808.30266131 · pubmed ↗
- 5M. Ciotti , S. L. Werlin , and I. S. Owens , “Delayed Response to Phenobarbital Treatment of a Crigler‐Najjar Type II Patient With Partially Inactivating Missense Mutations in the Bilirubin UDP‐Glucuronosyltransferase Gene,” Journal of Pediatric Gastroenterology and Nutrition 28 (1999): 210–213, 10.1097/00005176-199902000-00024.9932859 · doi ↗ · pubmed ↗
- 6M. A. Rahman , M. S. U. Islam , and N. Tasnim , “Crigler Najjar Syndrome Type 2: A Case of Unexplained Jaundice in an Adult,” Faridpur Medical College Journal 15 (2020): 43–45, 10.3329/fmcj.v 15i 1.49011. · doi ↗
- 7CARE Case Report Guidelines [Internet] , “CARE Case Rep. Guidel,” https://www.care‐statement.org.
- 8K.‐H. Wagner , R. G. Shiels , C. A. Lang , N. Seyed Khoei , and A. C. Bulmer , “Diagnostic Criteria and Contributors to Gilbert's Syndrome,” Critical Reviews in Clinical Laboratory Sciences 55 (2018): 129–139, 10.1080/10408363.2018.1428526.29390925 · doi ↗ · pubmed ↗